


Hereditary angio-oedema (HAE) is manifested by repeated episodes of localised subcutaneous or sub-mucosal oedema. Symptoms are extremely variable in frequency, localisation, and severity. Atypical or mild clinical symptoms of the disease may lead to erroneous diagnosis, causing diagnostic delay. The goal of this study was to assess how diagnostic delay has changed over 33 years at a single referral centre.
MethodsWe analysed diagnostic delay and first symptoms of HAE in patients who were diagnosed at an immunology department between 1980 and 2013. Patient's records were analysed.
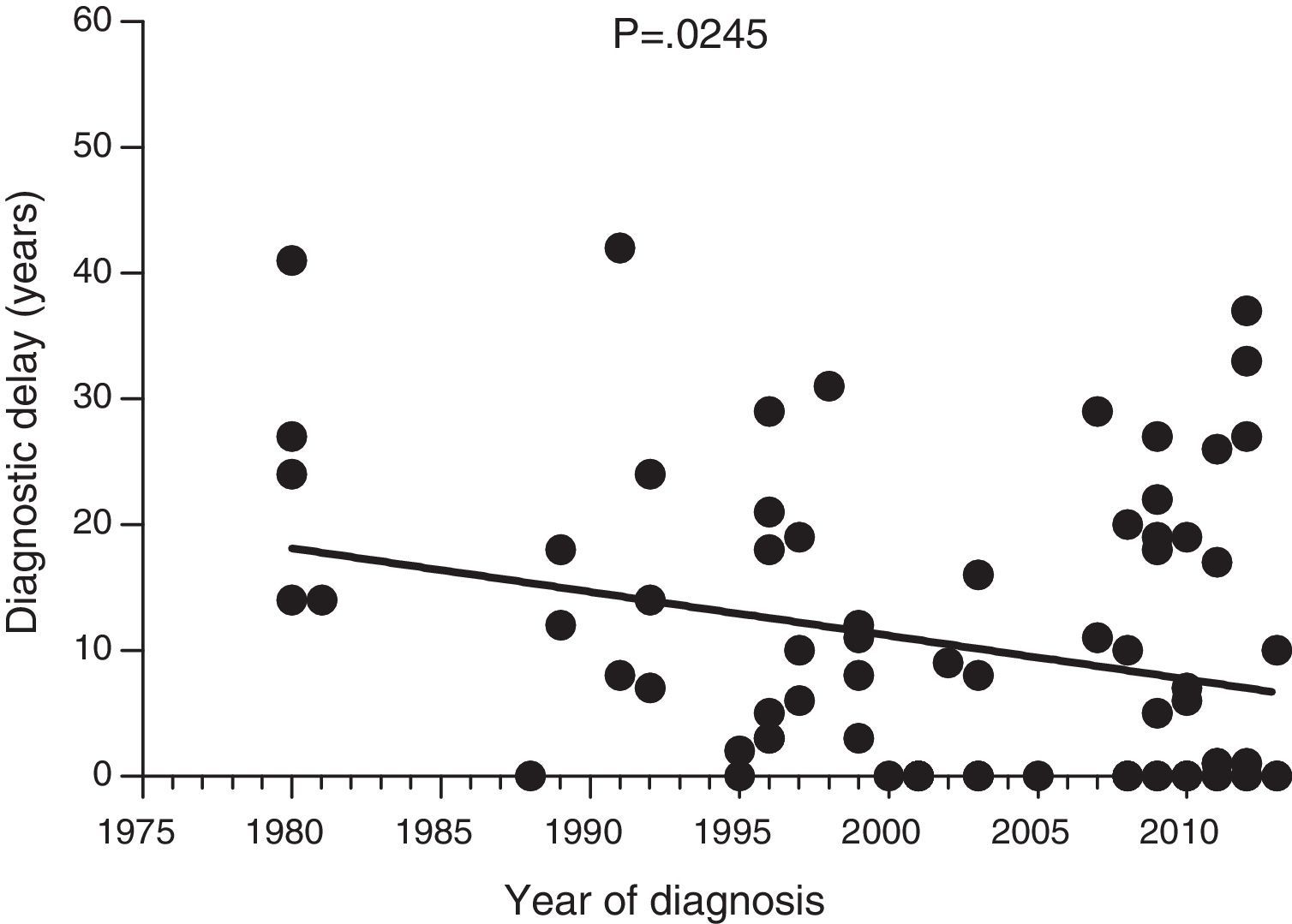
ResultsThe median diagnostic delay in 77 HAE type 1 and 2 patients was seven (range, 0–42) years. The difference observed in diagnostic delay between probands (18 [0–42] years) and others (1 [0–37] year) was significant (p<0.001). Our data show a significant negative correlation between the length of diagnostic delay and the year of diagnosis in our group of patients (p=0.024). The median age of first symptoms among all HAE patients (N=64) was 17 (1–40) years. The first symptoms of HAE in 64 patients were analysed. Twenty-six patients had abdominal, seventeen peripheral, five facial, two urogenital, and three had laryngeal oedema as the first manifestation of the disease. The last death that was attributed to HAE was in 1977.
ConclusionsOur observations demonstrate improved awareness of HAE among physicians, as documented by the significant decrease in diagnostic delay. It is believed that earlier treatment will improve patient quality of life and life expectancy.
Hereditary angio-oedema (HAE) is a rare disease that is caused by C1 inhibitor (C1-INH) deficiency, with an estimated frequency of 1 in 50,000 in the general population and no major ethnic or gender differences in prevalence.1,2 The C1-INH protein is a serine protease inhibitor that is important for regulating the activation of the complement system, the contact system, and the intrinsic coagulation system. During HAE attacks, plasma proteolytic cascades are activated, and several vasoactive substances are generated.1 Studies have shown that bradykinin is the predominant mediator of enhanced vascular permeability in HAE attacks.3,4 Clinically, HAE is manifested by repeated episodes of localised subcutaneous or submucosal oedema. Symptoms of HAE are extremely variable in frequency, localisation, and severity. Clinical signs of the disease may lead to erroneous diagnosis, causing diagnostic delay. Possible provoking factors of HAE attacks include mechanical trauma, stress, infection, menstruation, pregnancy, surgical or diagnostic intervention, and certain drugs (e.g., ACE inhibitors or oestrogen medications).5,6 The disease is inherited as an autosomal dominant trait. The spontaneous mutation rate is about 25%; more than 100 C1-INH gene mutations (SERPING1) have been described.7 Delay in diagnosis is common in patients with hereditary angio-oedema. As of 1977, the average time between the onset of symptoms and diagnosis was 22 years, and in 2005 it was still greater than 10 years.1 Even today, the diagnostic delay is markedly different between European countries, ranging from 2.0 years in Germany to 16.3 years in Denmark.8,9 Delayed treatment of laryngeal oedema can result in death. The goal of this study was to evaluate how the diagnosis of HAE has changed in a single referral centre during the last 33 years.
MethodsA retrospective analysis of the records of every patient ever diagnosed with HAE at the Department of Clinical Immunology and Allergology in Brno, Czech Republic was performed. The department is a referral centre for Moravia (the eastern part of the Czech Republic), which has a population of 4,124,774. The centre was established in 1980, when the first four patients were diagnosed. All of the data on our patients were collected from the medical records by clinicians. Four main areas in the medical records were searched: demographics, diagnosis and diagnostic delay, first symptoms, and family history.
Over the last 33 years, methods for detecting C3, C4 and C1-INH levels have changed in our department. In the 1980s and 1990s these levels were detected by radial immunodiffusion and consequently by nephelometry. Since 2006, IMMAGE Beckman coulter nephelometry has been used. Functional assays, specifically the C1 inhibitor Enzyme-Linked ImmunoSorbent Assay (Quidel MicroVue C1 Inhibitor Plus), have been used since 1996. The normal range for C1-INH concentration is 210–390mg/L, and the normal range for functional activity is greater than 68% of the reference value for standard serum. Type I HAE is diagnosed when both the C1-INH concentration and function are below normal limits. Type II HAE is diagnosed when C1-INH concentration is normal or high, but the function level is below normal reference value ranges. Diagnosis of HAE is supported by a low C4 level.
This project was approved by St Anne's University Hospital Ethics Committee.
Statistical analysisTo evaluate diagnostic delay, the Mann–Whitney test and Spearman's rank correlation test were used. Categorical variables were analysed using Fisher's exact test. A standard level of statistical significance α=0.05 was used. If not otherwise indicated, results are given as a median (range). The statistical packages GraphPad Prism 5 and Statistica 12 were used.
ResultsCharacteristics of the patientsBetween 1980 and 2013, 77 HAE patients from 31 families (i.e., with 31 probands) were diagnosed. In six patients HAE did not affect other known family members. In 42 patients from 31 families, HAE was genetically confirmed by molecular genetic analysis. Two patients died: one of stroke in 2009 (aged 61 years) and one of pulmonary fibrosis in 2006 (aged 38 years). Three moved to other regions of the Czech Republic. Our study population consisted of 61 patients with HAE type 1 (33 female) and 16 with HAE type 2 (10 female). These data show that the estimated prevalence of HAE in the Moravian region is 1:54,000. In 44 patients (69%), the first manifestation of the disease was before the age of 20, while in only two (3%), manifestation began after 30 years of age. Twenty-six patients (34%) were diagnosed with type 1 or 2 HAE during family screening prior to the onset of clinical symptoms.
Symptoms at first manifestationTwenty-six (40.6%) of the 64 symptomatic patients experienced abdominal oedema as the first manifestation of HAE. Seventeen patients (26.6%) experienced peripheral skin oedema and two (3.1%) experienced urogenital oedema. Three patients (4.7%) experienced laryngeal oedema and were aged 12, 18, and 21 years at first manifestation of the disease; all were diagnosed between 1980 and 2000. Eleven patients (17%) experienced a combination of peripheral skin, abdominal, urogenital, and laryngeal oedemas at the first manifestation of HAE. Between 1980 and 2000, 30 symptomatic patients were diagnosed. No asymptomatic patient was diagnosed in that period. Between 2001 and 2013, there were 47 patients diagnosed, 13 of whom were asymptomatic (p=0.007, Fischer's exact test).
Age of first symptomsThe median age of the first symptoms was 17 (1–40) years. Among HAE type 1 patients (n=50), the median age was 17.5 (1–40) years, whereas among HAE type 2 patients (n=14), median age was significantly lower at 11 (3–26) years (p=0.046, Fischer's exact test).
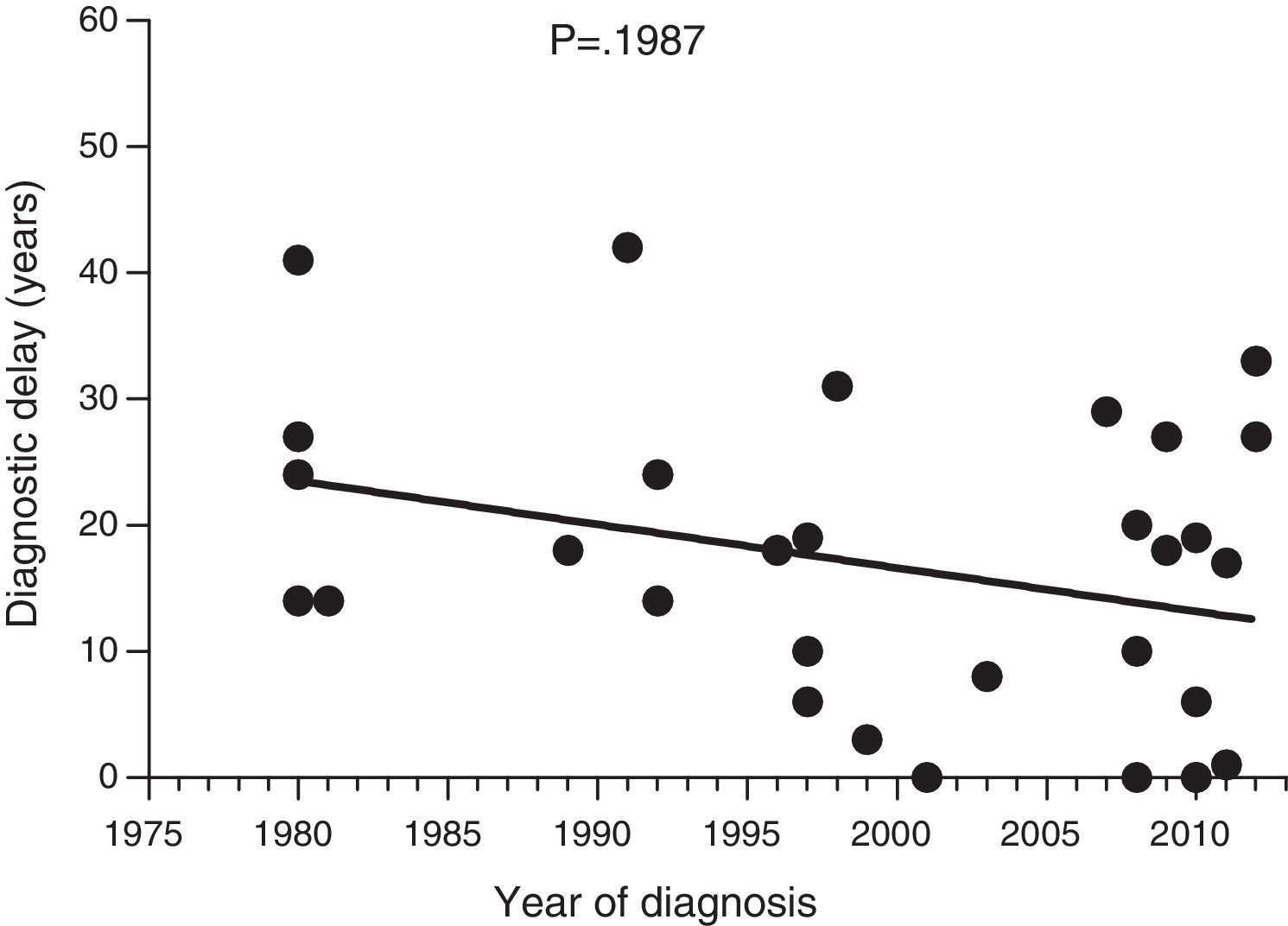
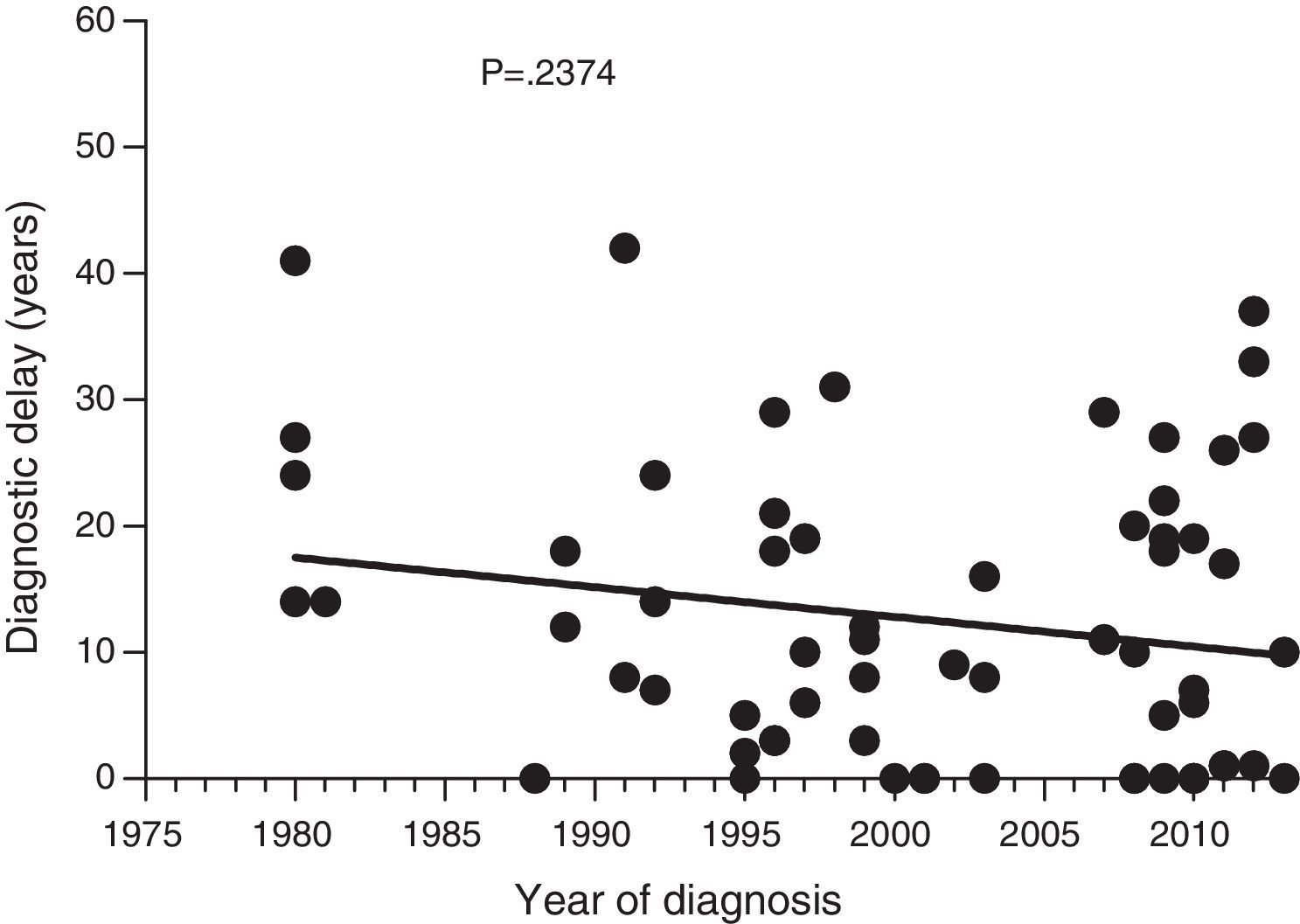
Diagnostic delayFig. 1 shows a significant negative correlation between the length of diagnostic delay and the year of diagnosis in our group of patients (r=−0.2562; p=0.024). However, no correlation was observed when the group of patients was divided into symptomatic patients (p=0.237) and probands (p=0.198) (Figs. 2 and 3).
. Spearman")
. Spearman")
. Spearman")
The median length of diagnostic delay among 77 HAE type 1 and 2 patients was seven (0–42) years. The difference observed in diagnostic delay between probands (18 [0–42] years) and other patients (1 [0–37] year) was significant (p<0.001, Mann–Whitney test). There were 13 asymptomatic patients (11 with type I HAE; p=0.270, Fischer's exact test) during the study. There was no difference in diagnostic delay between HAE type 1 and type 2 patients (p=0.084).
Death associated with HAEThree families recalled a total of three relatives (all women) who died of suffocation during a probable HAE attack. In each of these families, type 1 HAE was diagnosed between 1980 and 2000, and the deaths occurred in 1956, 1960, and 1977. In all cases the death occurred before HAE was diagnosed. A possible trigger factor was identified in one woman only, when a fatal laryngeal attack followed tooth extraction. We can hypothesise that the lack of death was due to increased awareness about HAE and the easy access by patients to treatment options, however direct evidence is not available.
Laryngeal oedema in HAE patientsIn our series, 11% (7 of 64) of patients had one or more episodes of laryngeal oedema. The laryngeal oedemas occurred between the 10th and 40th years of life. However, no death was associated with laryngeal oedema.
DiscussionThe goal of this study was to assess how diagnostic delay has changed during the last 33 years at a single referral centre. We demonstrate that the median diagnostic delay was seven (0–42) years in our setting. Zanichelli et al.9 analysed diagnostic delay in eight countries and found a median delay of 8.5 (0–62) years. The diagnostic delay was markedly different between countries, ranging from 2.0 (0–62) years in Germany to 15 (0–57) years in Italy. However, no statistically significant differences existed, even when analysed by year of diagnosis.9 National surveys in Spain and Denmark have reported mean diagnostic delays of 13.1 and 16.3 years, respectively.8,10
Data collected on diagnostic delay in the 77 patients in our study group showed a significant negative correlation between the length of diagnostic delay and the year of diagnosis. To our knowledge, this is the first documented decrease in diagnostic delay in HAE patients. However, this decrease was not significant for symptomatic patients; the decrease is, at least partly, attributed to early diagnosis of asymptomatic patients.
The insignificantly shorter diagnostic delay seen between HAE 1 and HAE 2 patients corresponds to findings observed in Danish and UK surveys.8,11 Several authors explain this fact by the different availabilities of functional C1-INH tests.8,9,11 In prior studies, patients were tested only for levels of C1 inhibitor, which are normal or elevated in HAE type 2 patients.9 In our setting, tests for level and function of C1 inhibitor were simultaneously performed together with C4 determination, which is highly sensitive for diagnosing both HAE type 1 and HAE type 2.
Compared to previous European studies, the mean age of symptom onset in our patients is higher (17 years). Zanichelli et al.9 analysed age at first symptoms and found that it was 12 years among HAE type 1 patients and 13 years among HAE type 2 patients. In Denmark, it was 9.5 years.8 In contrast, in China it was 21.3 years, whereas in Brazil, it was 6.5 years.12,13
Between 1980 and 2013, no deaths in our setting were attributed to HAE. The last probable case was recorded in 1977. In Denmark between 1886 and 2009, at least 11 HAE-related deaths were described by relatives for 82 patients.8 Possible reasons for this difference include the availability of C1 inhibitor therapy in our region since the 1980s for the treatment of acute attacks and the establishment of long-term prophylaxis with attenuated androgens and relatively good awareness about the disease, primarily among allergists. In our setting, laryngeal oedema was observed as the first symptom in only three patients (4.68%), which agrees with similar observations in Germany.14
The most frequently observed first manifestation of HAE was abdominal attacks, followed by peripheral oedemas. In Denmark, 60 patients (78% of the symptomatic patients) could recall the location of their first attack. Twenty-nine mentioned a skin oedema, and 28 mentioned an abdominal attack. Three patients had their first swelling in the mouth area.8
In our series, 11% of patients had one or more episodes of laryngeal oedema. This is markedly less than the levels seen in other European countries. In the Italian study, 48% of 226 HAE patients experienced episodes of laryngeal oedema.15 American and Italian studies found rates of 50% (45 of 89 patients)16 and 78% (81 of 104 patients),17 respectively.
A family history of HAE can lead to consultation with a physician and to a medical evaluation that is focused on HAE. In this study 33.8% of patients were diagnosed as a result of familial incidence. Compared to a similar analysis carried out in the UK and Brazil, a larger percentage of our patients were diagnosed prior to the onset of first symptoms because of family incidence. In a national audit of hereditary and acquired angioedema in the UK, 3% were found as a result of family incidence, and in Brazil it was 13.8%.11,12
In our retrospective study, we analysed relevant data from one centre over a 33-year period and found that diagnostic delay was reduced. There was a trend towards decreased diagnostic delay in our symptomatic patients; however this was not significant and may be partially attributed to improved diagnosis of the affected offspring of previously diagnosed symptomatic probands. Our centre has not recorded any deaths associated with undiagnosed HAE since 1977. It is supposed that earlier diagnosis means earlier treatment, which will improve the patient quality of life and possibly also life expectancy.
Ethical disclosuresProtection of human subjects and animals in researchProtection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
This report was written at Masaryk University as part of the project “Některé laboratorní a klinické aspekty humorálních imunodeficiencí” number MUNI/A/1182/2014 with the support of the Specific University Research Grant, as provided by the Ministry of Education, Youth and Sports of the Czech Republic in the year 2015. This work was also supported by the grant 15-28732A of the Czech Health Research Council.
