




El tratamiento de la tuberculosis ha de ser prolongado, ya que existen poblaciones de bacilos diferentes en cuanto a su actividad metabólica, y debe asociar varios fármacos, puesto que tienen lugar mutaciones cromosómicas espontáneas que producen resistencia que se seleccionaría con la monoterapia. El fenotipo multirresistente se debe a la adquisición secuencial de mutaciones en diferentes loci independientes. Los principales desafíos en el tratamiento de la tuberculosis son, por una parte, las cepas resistentes y, por otra, conseguir acortar la duración y dosificación del tratamiento para facilitar la adherencia a éste. Para esto, se precisan fármacos que actúen rápidamente, que sean activos frente a las cepas resistentes y que sean capaces de eliminar las poblaciones “durmientes”. En los últimos años se han desarrollado análogos de los fármacos de primera línea con una dosificación más larga (rifamicinas) o que pueden reducir la duración del tratamiento (moxifloxacino, gatifloxacino). En cualquier caso, son necesarios nuevos fármacos con mecanismos de acción diferentes (sin resistencias cruzadas con los demás antituberculosos), y que tengan el potencial de reducir la duración y la frecuencia de dosificación del tratamiento. En esta revisión se describen las características, la actividad, los mecanismos de resistencia y los efectos secundarios de los distintos antituberculosos.
Treatment for Mycobacterium tuberculosis must be lengthy because populations of this bacillus differ in metabolic activity, and it must include various associated drugs, since spontaneous chromosome mutations can give rise to drug resistance. The multiresistant phenotype emerges with sequential acquisition of mutations in several independent loci. Thus, the main challenges in tuberculosis treatment are resistant strains and reductions in treatment duration and dose to facilitate adherence on the part of the patient. The drugs should be fast acting, active against resistant strains, and able to eradicate slowly metabolizing populations. Progress in recent years has led to the development of analogs of first-line antituberculosis drugs with longer dosing intervals (rifamycin derivatives) or shorter treatment courses (moxifloxacin, gatifloxacin). Nonetheless, new drugs that additionally have novel mechanisms of action and no cross-resistance to current first-line drugs are needed. This review describes the characteristics, activity, resistance mechanisms, and side effects associated with antituberculosis drugs.
El tratamiento de la tuberculosis se basa en conceptos muy distintos a los de las demás infecciones bacterianas. Mycobacterium tuberculosis tiene un tiempo de generación prolongado y la capacidad para entrar en períodos de latencia con una actividad metabólica limitada, lo que dificulta la acción de los antimicrobianos1. Existen poblaciones de bacilos diferentes en función de su localización y actividad. Así, los bacilos presentes en las cavidades pulmonares se multiplican de forma activa en un ambiente aerobio, los del interior de los macrófagos lo hacen en un ambiente microaerofílico que induce la latencia y los que se encuentran en el interior del caseum tienen sólo ocasionalmente un ciclo replicativo (crecimiento intermitente). Por otra parte, M. tuberculosis puede multiplicarse en los tejidos, donde la penetración de los antibióticos es fácil, o bien encontrarse en cavidades pulmonares, pus o material caseoso, en donde la penetración de los antibióticos resulta más difícil. Finalmente, hay que señalar que el pH del material caseoso y el del interior de los macrófagos es muy bajo, lo que condiciona la actividad de los distintos fármacos2. Los fármacos antituberculosos presentan un perfil de actividad diferenciado frente a cada una de estas localizaciones y poblaciones, y es necesario asegurarse de que el tratamiento prescrito sea activo frente a todas ellas.
Las micobacterias presentan una resistencia natural a numerosos antibacterianos, por el hecho de poseer una pared compleja, muy hidrófoba, con una permeabilidad reducida para un gran número de compuestos3; por esto, el tratamiento de la tuberculosis se realiza con antimicrobianos específicos (con actividad antituberculosa). También se han descrito enzimas modificantes como las betalactamasas4 o sistemas de eflujo5.
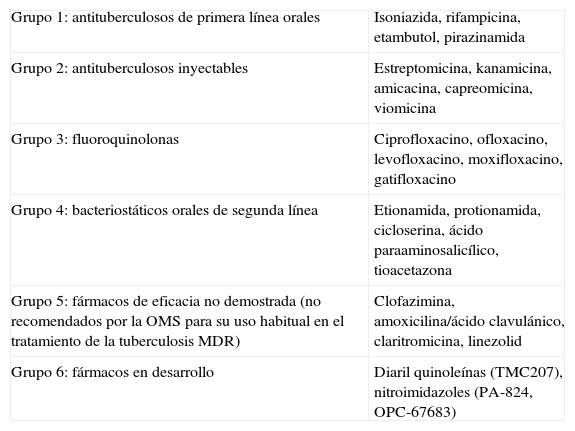
La tuberculosis tiene un tratamiento eficaz, basado en la asociación de fármacos y una larga duración, que asegura en los casos no complicados una tasa de curación superior al 95%, siempre que se complete. El esquema terapéutico recomendado por la OMS (DOTS [directly observed treatment, short course]) incluye isoniazida (INH), rifampicina (RIF), etambutol (ETB) y pirazinamida (PZA) los primeros 2 meses e INH y RIF hasta completar los 6 meses de tratamiento. En los niños, se sustituirá el ETB por estreptomicina (STR), ya que no es posible monitorizar la agudeza visual. Los fármacos mencionados se conocen como fármacos de primera línea para el tratamiento de la tuberculosis. Existe cierto debate en España sobre la incorporación del cuarto fármaco (ETB o STR) en el tratamiento inicial, dada la baja tasa de resistencia a la INH en enfermos sin tratamiento previo6. En cualquier caso, el cuarto fármaco sería como mínimo necesario cuando el paciente se ha tratado previamente con antimicobacterianos, cuando procede de un país con elevada resistencia primaria a la INH (>4%) o cuando existe antecedente de contacto con un paciente con tuberculosis resistente conocida. Existen también otros fármacos de segunda línea o de reciente desarrollo que constituyen, como se verá, alternativas para el tratamiento (tablas 1 y 2).
Clasificación de los fármacos antituberculosos1
| Grupo 1: antituberculosos de primera línea orales | Isoniazida, rifampicina, etambutol, pirazinamida |
| Grupo 2: antituberculosos inyectables | Estreptomicina, kanamicina, amicacina, capreomicina, viomicina |
| Grupo 3: fluoroquinolonas | Ciprofloxacino, ofloxacino, levofloxacino, moxifloxacino, gatifloxacino |
| Grupo 4: bacteriostáticos orales de segunda línea | Etionamida, protionamida, cicloserina, ácido paraaminosalicílico, tioacetazona |
| Grupo 5: fármacos de eficacia no demostrada (no recomendados por la OMS para su uso habitual en el tratamiento de la tuberculosis MDR) | Clofazimina, amoxicilina/ácido clavulánico, claritromicina, linezolid |
| Grupo 6: fármacos en desarrollo | Diaril quinoleínas (TMC207), nitroimidazoles (PA-824, OPC-67683) |
MDR: multirresistente; OMS: Organización Mundial de la Salud.
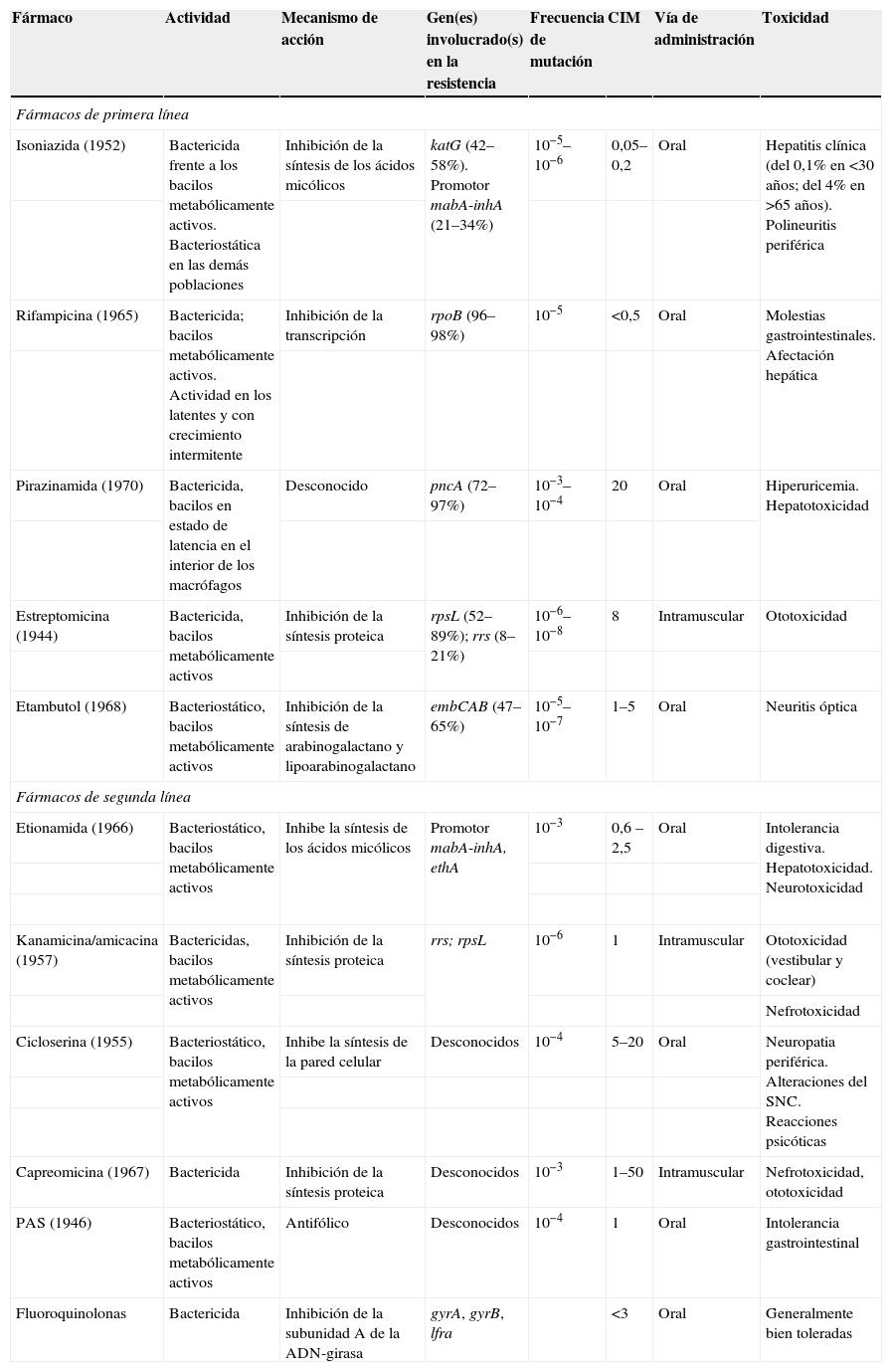
Características de los fármacos antituberculosos
| Fármaco | Actividad | Mecanismo de acción | Gen(es) involucrado(s) en la resistencia | Frecuencia de mutación | CIM | Vía de administración | Toxicidad |
| Fármacos de primera línea | |||||||
| Isoniazida (1952) | Bactericida frente a los bacilos metabólicamente activos. Bacteriostática en las demás poblaciones | Inhibición de la síntesis de los ácidos micólicos | katG (42–58%). Promotor mabA-inhA (21–34%) | 10−5–10−6 | 0,05–0,2 | Oral | Hepatitis clínica (del 0,1% en <30 años; del 4% en >65 años). Polineuritis periférica |
| Rifampicina (1965) | Bactericida; bacilos metabólicamente activos. Actividad en los latentes y con crecimiento intermitente | Inhibición de la transcripción | rpoB (96–98%) | 10−5 | <0,5 | Oral | Molestias gastrointestinales. Afectación hepática |
| Pirazinamida (1970) | Bactericida, bacilos en estado de latencia en el interior de los macrófagos | Desconocido | pncA (72–97%) | 10−3–10−4 | 20 | Oral | Hiperuricemia. Hepatotoxicidad |
| Estreptomicina (1944) | Bactericida, bacilos metabólicamente activos | Inhibición de la síntesis proteica | rpsL (52–89%); rrs (8–21%) | 10−6–10−8 | 8 | Intramuscular | Ototoxicidad |
| Etambutol (1968) | Bacteriostático, bacilos metabólicamente activos | Inhibición de la síntesis de arabinogalactano y lipoarabinogalactano | embCAB (47–65%) | 10−5–10−7 | 1–5 | Oral | Neuritis óptica |
| Fármacos de segunda línea | |||||||
| Etionamida (1966) | Bacteriostático, bacilos metabólicamente activos | Inhibe la síntesis de los ácidos micólicos | Promotor mabA-inhA, ethA | 10−3 | 0,6 –2,5 | Oral | Intolerancia digestiva. Hepatotoxicidad. Neurotoxicidad |
| Kanamicina/amicacina (1957) | Bactericidas, bacilos metabólicamente activos | Inhibición de la síntesis proteica | rrs; rpsL | 10−6 | 1 | Intramuscular | Ototoxicidad (vestibular y coclear) |
| Nefrotoxicidad | |||||||
| Cicloserina (1955) | Bacteriostático, bacilos metabólicamente activos | Inhibe la síntesis de la pared celular | Desconocidos | 10−4 | 5–20 | Oral | Neuropatia periférica. Alteraciones del SNC. Reacciones psicóticas |
| Capreomicina (1967) | Bactericida | Inhibición de la síntesis proteica | Desconocidos | 10−3 | 1–50 | Intramuscular | Nefrotoxicidad, ototoxicidad |
| PAS (1946) | Bacteriostático, bacilos metabólicamente activos | Antifólico | Desconocidos | 10−4 | 1 | Oral | Intolerancia gastrointestinal |
| Fluoroquinolonas | Bactericida | Inhibición de la subunidad A de la ADN-girasa | gyrA, gyrB, lfra | <3 | Oral | Generalmente bien toleradas | |
ADN: ácido desoxirribonucleico; CIM: concentración inhibitoria mínima; PAS: ácido paraaminosalicílico; SNC: sistema nervioso central.
El médico responsable deberá asegurarse de la adherencia al tratamiento, que puede verse dificultada por el coste, los efectos secundarios o su duración. La falta de adherencia ha contribuido a la aparición de cepas resistentes, multirresistentes (MDR) y extremadamente resistentes (XDR). Se estima que en el año 2006 hubo 489.139 casos nuevos de tuberculosis por cepas MDR (4,8% del total). Las zonas con mayor incidencia de tuberculosis MDR fueron China, la India y los países de la antigua Unión Soviética. Estas cepas son mucho menos frecuentes en América, Europa central y occidental y África7.
La paradoja del tratamiento de la tuberculosis es que cuando se cumplen 50 años de la introducción de una quimioterapia eficaz: el número de casos de la enfermedad a escala mundial es mayor y, lo que es más alarmante, aumenta el número de infecciones por cepas resistentes, MDR y XDR a los fármacos antituberculosos. Así, en el año 2006 existían más de 1.500 millones de infectados (un tercio de la población mundial), aproximadamente 9,2 millones de nuevos casos de la enfermedad cada año y 1,7 millones de muertes8. A esta situación han contribuido la pandemia por el virus de la inmunodeficiencia humana (VIH), la reducción de la financiación de los programas de control de la enfermedad, los movimientos migratorios a partir de países con gran incidencia de tuberculosis y el incremento de la incidencia de cepas MDR. Por esto, en 1993, la OMS declaró la tuberculosis como una emergencia de salud pública global.
Estudios genéticos han demostrado que la resistencia a los fármacos antituberculosos se debe a mutaciones cromosómicas espontáneas de los genes que codifican la diana del fármaco o a enzimas implicadas en la activación del fármaco9–11. Se han descrito mutaciones puntuales, deleciones o inserciones responsables de la resistencia a fármacos antituberculosos de primera línea o a algunos de segunda línea. La tasa con la que tienen lugar estas mutaciones difiere para cada uno de los antimicrobianos; la más elevada es la del ETB y la más baja, la de la RIF y las quinolonas. No obstante, no se conoce una alteración genética que, por sí misma, dé lugar al fenotipo de multirresistencia (definido como la asociación de resistencias como mínimo a INH y a RIF). Este fenotipo MDR se debe a la adquisición secuencial de mutaciones en diferentes loci de genes independientes12. La detección precoz de la resistencia a los fármacos antituberculosos es esencial para el correcto control de la tuberculosis resistente13. El conocimiento de los mecanismos de resistencia permite desarrollar técnicas moleculares para su detección precoz14–16.
Antimicobacterianos de primera líneaIsoniazidaLa INH es un fármaco sintético introducido en terapéutica en 1952. Los miembros del complejo tuberculoso son, generalmente, muy sensibles a la INH, mientras que el papel de la INH en el tratamiento de otras micobacterias es muy limitado.
De hecho, la INH es un profármaco que requiere una activación in vivo que producirá un potente derivado, capaz de oxidar o acilar grupos proteicos, que finalmente actuará en la síntesis de los ácidos micólicos de la pared de M. tuberculosis. La activación de la INH se lleva a cabo por la enzima catalasa-peroxidasa codificada por el gen katG. En el 42 al 58% de las cepas resistentes a la INH se encuentran mutaciones del gen katG9,10. A pesar de que se ha descrito un número importante de mutaciones de este gen, la más frecuente es la Ser315Thr, que se encuentra en aproximadamente el 40% de las cepas resistentes. La enzima resultante es incapaz de activar la INH y provoca una resistencia de alto nivel (concentración inhibitoria mínima [CIM] superior a 1mg/ml), pero mantiene el 50% de su actividad catalasa-peroxidasa, asegurando un nivel de protección oxidativa suficiente17. La ausencia o disminución de la actividad catalásica en las cepas de M. tuberculosis resistentes a la INH se conoce desde 195418.
Si la enzima catalasa-peroxidasa se encuentra intacta, el derivado activo de la INH actúa bloqueando la síntesis de los ácidos micólicos. Una de las dianas intracelulares del derivado activo sería la reductasa de la proteína transportadora de ácidos grasos acilenólicos codificada por el gen inhA19. En aproximadamente el 25% de las cepas resistentes se producen mutaciones en la región inhA y suelen asociarse a una resistencia de bajo nivel. La mayor parte de estas mutaciones se han descrito en la región reguladora del gen y originan una sobreexpresión de la enzima que compensa la acción inhibidora del fármaco20. Ocasionalmente se han descrito mutaciones en la zona de interacción de la enzima y la INH. Del 10 al 20% de las cepas resistentes a la INH no presenta alteraciones del gen katG ni del inhA. Se han investigado otros genes, como el ahpC (alquil hidroperoxidorreductasa), el kasA (cetoácido sintasa), el ceoA (uridin difosfato fenol [UDP] galactopiranosa reductasa) y las deshidrogenasas de malato y nicotinamida-adenina dinucleótido reducido10. El papel exacto de las alteraciones encontradas en estos genes respecto a la resistencia a la INH queda por determinar.
La INH es activa tan sólo frente a los bacilos en replicación activa; su papel es muy limitado en las poblaciones que replican lentamente, como las del caseum, o en las poblaciones latentes, como las del interior de los macrófagos. La mutación espontánea que produce la resistencia se da con una frecuencia21 de 10−5 a 10−6. La CIM en las cepas sensibles oscila entre 0,025 y 0,05μg/ml. Las concentraciones séricas después de 2h de una dosis oral de 300mg son de 4,5 a 7μg/ml. Puede producir hepatotoxicidad y se ha descrito elevación de las aminotransferasas subclínica entre el 10 y el 15% de los pacientes tratados. El riesgo de la hepatitis clínica es muy bajo antes de los 30 años (0,1%) y se incrementa de manera progresiva hasta el 4% en mayores de 65 años22. También puede originar una polineuritis periférica por déficit de piridoxina. Los efectos neurológicos de la INH pueden prevenirse con una dosis diaria de 10 a 50mg/día de piridoxina, aunque sólo se utilizará cuando existan factores de riesgo (diabetes, alcoholismo, embarazo, malnutrición o infección por VIH).
RifampicinaLa RIF, rifamicina SV con un grupo aminometilpiperacina en posición 3, es un antimicrobiano semisintético introducido en terapéutica en el año 1967. La RIF, al igual que las demás rifamicinas, es un potente inhibidor de la síntesis de ARN mensajero (ARNm) y, por tanto, de la transcripción genética, al unirse a la polimerasa del ARN dependiente del ADN de los procariotas. En Escherichia coli, esta enzima es un oligómero compuesto por 4 subunidades distintas (α, β, β‘ y σ, que están codificadas por los genes rpoA, rpoB, rpoC y rpoD, respectivamente). La inhibición de la transcripción tiene lugar porque la RIF se fija a la subunidad β23. Además de un efecto bactericida sobre las células de M. tuberculosis metabólicamente activas, la RIF también posee acción esterilizante de las bacterias en estado de latencia (que sólo presentan ocasionalmente actividad metabólica), tanto en los focos necróticos como en el interior de los macrófagos. Esta actividad de la RIF, junto con la inclusión de la PZA en los esquemas terapéuticos, ha permitido acortar el tratamiento de la tuberculosis no complicada a 6 meses.
La mutación espontánea que origina la resistencia se observa con una frecuencia21 de 10−8. Las CIM para M. tuberculosis son muy bajas (0,005–0,2μg/ml). El pico sérico a las 2–4h de una dosis oral de 600mg es de 5–10μg/ml. Puede producir molestias gastrointestinales (dolor epigástrico, anorexia, náuseas, vómitos y diarrea) y afectación hepática (en menos del 1,1% de los pacientes). También desencadena reacciones de hipersensibilidad, como fiebre, prurito, urticaria, vasculitis cutánea, eosinofilia, trombocitopenia, hemólisis o insuficiencia renal por nefritis intersticial, que son más frecuentes en los pacientes con infección por VIH y tratamiento intermitente. Tiñe la orina, el sudor y las lágrimas de color naranja. Es un potente inductor enzimático, puede disminuir las concentraciones séricas de otros fármacos administrados concomitantemente, como, entre otros, antirretrovirales inhibidores de la proteasa, anticonceptivos orales, anticoagulantes orales, digoxina, corticoides, ciclosporina, tacrolimus y sulfonilurea.
Las micobacterias, al igual que otros géneros bacterianos, adquieren resistencia a la RIF mediante mutaciones en una región bien definida de 81 pb (27 codones) de la región central del gen que codifica para la subunidad β de la polimerasa del ARN (rpoB). Más del 96% de las cepas resistentes a la RIF posee mutaciones en esta región24,25. Este hecho facilita enormemente el desarrollo de técnicas moleculares para la detección rápida de la resistencia a la RIF en la cepa aislada por cultivo o, incluso, directamente en la muestra clínica26. A pesar de que se han descrito 35 variantes alélicas, con ligeras variaciones en su distribución geográfica, las mutaciones más comunes (65–86%) son las que afectan al codón 526 o al codón 531 y dan lugar a una resistencia de alto nivel (CIM >32μg/ml). Hay que señalar que en el 4% de las cepas no se detectan mutaciones en este fragmento de 81 pb. En estas cepas, la resistencia puede deberse a mutaciones fuera de esta región, que se han descrito particularmente en la región aminoterminal9, o a mecanismos alternativos, como alteraciones en la permeabilidad a la RIF25. Así como la resistencia aislada a la INH se observa con cierta frecuencia, la resistencia a la RIF se asocia a la resistencia a la INH en más del 95% de los casos, por lo que su detección constituye un marcador de multirresistencia. Ocasionalmente se ha descrito monorresistencia a la RIF27.
PirazinamidaLa PZA es un derivado sintético de la nicotinamida. Posee un potente efecto esterilizante sobre los bacilos tuberculosos latentes en el interior de los macrófagos, que permite, cuando se usa asociada a la RIF, acortar la duración del tratamiento de la tuberculosis no complicada a 6 meses. Carece de actividad frente a las demás micobacterias, incluidos otros componentes del complejo tuberculosis que son constitutivamente resistentes a la PZA debido a una mutación puntual del gen pncA (C→G en el codón 169). La PZA es un profármaco que la enzima micobacteriana pirazinamidasa convierte a su forma activa, el ácido pirazinoico. La PZA se difunde pasivamente al interior de los macrófagos, donde se convierte en ácido pirazinoico que se acumula intracelularmente por un sistema ineficiente de bombeo. El ácido pirazinoico actúa sobre su diana, una enzima implicada en la síntesis de los ácidos micólicos28, tanto directamente como porque disminuye el pH intracelular por debajo de los límites tolerados por esta enzima.
La CIM promedio de M. tuberculosis es de 20μg/ml. Entre una y 4h después de una dosis oral de 1g se obtienen picos séricos de alrededor de 45μg/ml. Durante el tratamiento se puede observar hiperuricemia por bloqueo de la secreción tubular renal de uratos, aunque suele ser subclínica, con la excepción de los enfermos gotosos. Ocasionalmente provoca hepatotoxicidad29 (los esquemas terapéuticos actuales con dosis de 20 a 35mg/kg/día son mucho menos hepatotóxicos) y alteraciones gastrointestinales. Excepcionalmente pueden aparecer exantema y fotosensibilidad.
Se ha observado que entre el 72 y el 97% de las cepas de M. tuberculosis resistentes a la PZA posee mutaciones dispersas del gen estructural (pncA) o del promotor de la pirazinamidasa30. No obstante, existen cepas resistentes a la PZA, sin mutaciones del gen pncA, en las que esta resistencia se debe a otros mecanismos relacionados con la permeabilidad o el eflujo31.
EtambutolEl ETB, etilen-diamino-dibutanol, es un producto isómero dextrógiro derivado de la etilendiamina. Es activo frente a M. tuberculosis, con CIM de 1 a 5μg/ml, aunque esta actividad requiere el crecimiento activo de las células susceptibles. Tiene una actividad variable sobre las demás especies de micobacterias de crecimiento lento y su actividad es mucho menor en las especies de crecimiento rápido. El ETB inhibe de forma específica la biosíntesis de la pared micobacteriana. Así, la resistencia al ETB se asocia a cambios en una región genómica definida, el operón embCAB32, que codifica arabinosil transferasa relacionada con la síntesis de componentes de la pared celular, como el arabinogalactano y el lipoarabinomanano33. La resistencia de alto nivel (>20μg/ml) se debe a un proceso múltiple que incluye como primer paso una sobreexpresión de las proteínas Emb, para posteriormente añadirse mutaciones en la región EmbB o cambios adicionales en los niveles de expresión. Entre el 47 y el 69% de las cepas de M. tuberculosis resistentes presenta mutaciones en la región EmbB. En las cepas que no presentan mutaciones en la región EmbB la resistencia suele ser menor, con CIM inferior a 10μg/ml32. La resistencia en M. tuberculosis se observa con una frecuencia de 10−5.
El pico sérico a las 2–4h de una dosis oral de 1,5g es de 5μg/ml. Puede causar neuritis óptica, relacionada con la dosis y la duración del tratamiento. Deben darse instrucciones al enfermo para que comunique inmediatamente cualquier disminución de la agudeza visual o dificultad para distinguir los colores rojo y verde. Como se ha comentado anteriormente, debido a la dificultad de monitorizar la agudeza visual no se recomienda utilizar ETB para el tratamiento de la tuberculosis infantil.
EstreptomicinaLa STR es un antibiótico aminoglucósido que interfiere en la síntesis proteica bloqueando la traducción del ARNm, tanto en su inicio como en la incorporación de nuevos aminoácidos a la cadena polipeptídica. La CIM para las cepas sensibles oscila alrededor de 8μg/ml, y se obtienen picos séricos de 25 a 50μg/ml tras una a 2h de una dosis intramuscular de 1g. Los efectos secundarios más importantes son las alteraciones vestibulares y con menos frecuencia las auditivas. Se relacionan con la dosis y la duración del tratamiento y pueden ser irreversibles. La frecuencia con la que aparecen mutantes resistentes en M. tuberculosis es de 10−6. En las micobacterias, el mecanismo conocido de resistencia a los aminoglucósidos es por alteración de la diana sobre la que actúan (16sRNA) como consecuencia de mutaciones cromosómicas. Estas mutaciones afectan a los genes que codifican el 16S ARNr (rrs) y la proteína ribosómica S12 (rpsL). Es interesante recordar que la mayoría de eubacterias tienen varias copias del operón ARNr, mientras que M. tuberculosis sólo tiene una. Así, en M. tuberculosis, una mutación que origine cambios nucleotídicos ya puede provocar resistencia (comportamiento dominante)25. Las mutaciones que afectan al gen rpsL se dan en el 52–59% de las cepas resistentes y producen una resistencia de alto nivel (CIM >500μg/ml), mientras que las que afectan al gen rrs se observan en el 8–21% de las cepas y producen una resistencia de nivel intermedio (CIM de 50–500μg/ml)10,34. La mayor parte de las mutaciones de rrs se circunscriben a la región 530. Esta región interacciona con la proteína ribosómica S12. Existen diferentes teorías que relacionan la estructura conformacional de la región 530 con la fidelidad de la traducción34. En aproximadamente un tercio de las cepas resistentes existe un nivel de resistencia bajo (CIM de 25–50μg/ml) y no se detectan alteraciones en los genes rrs o rpsL. En estas cepas se ha evidenciado un mecanismo de permeabilidad para justificar la resistencia34.
Antimicobacterianos de segunda líneaÁcido paraaminosalicílicoEl ácido paraaminosalicílico (PAS) es activo frente a M. tuberculosis, mientras que los demás microorganismos, incluidas las demás micobacterias, son resistentes. Es activo frente a la población de crecimiento extracelular. El mecanismo de acción del PAS no se conoce con exactitud. Se le ha atribuido una inhibición de la síntesis de ácido fólico, del metabolismo del ácido salicílico y del transporte de hierro. La CIM para M. tuberculosis es de 1μg/ml, y se obtienen picos séricos de 7 a 8μg/ml después de 1 a 2h de una dosis de 4g. El ácido PAS se absorbe de forma incompleta en el tracto digestivo, por lo que la dosis oral es elevada (10–12g/día), lo que, unido a una cierta intolerancia gastrointestinal, puede llevar a problemas de cumplimiento terapéutico.
CicloserinaLa D-cicloserina (4-amino-3-isoxazolidinona) es un análogo de la D-alanina que inhibe de forma competitiva las enzimas D-alanil-D-alanina sintetasa, alanina racemasa y alanina permeasa, e interfiere con la síntesis de la pared micobacteriana51. Es activa frente a todas las micobacterias, así como frente a otros microorganismos. Las CIM para M. tuberculosis oscilan entre 5 y 20μg/ml, y se obtienen picos séricos de alrededor de 10μg/ml a las 4h de una dosis oral de 250mg. La resistencia a la cicloserina parece depender de alteraciones en la D-alanil-D-alanina sintetasa35. La toxicidad es muy elevada e incluye neuropatía periférica; alteraciones del sistema nervioso central, como confusión, irritabilidad, cefalea, disartria, vértigo o convulsiones y alteraciones psicóticas que incluyen la depresión grave con ideas suicidas. Por esto, no suele utilizarse cuando existen alternativas.
Etionamida, protionamidaLa etionamida (ETH) (etil-tio-isonicotinamida) y la protionamida (propil-tio-isonicotinamida) son derivados del ácido isonicotínico con una potente actividad frente a M. tuberculosis y otras micobacterias. La ETH inhibe la síntesis de ácidos micólicos y estimula las reacciones de oxidorreducción. Su mecanismo de acción es, pues, parecido al de la INH. De hecho, las mutaciones del gen inhA, que confieren una resistencia de bajo nivel a la INH, también producen resistencia a la ETH36. Hay que destacar, no obstante, que las cepas con resistencia de alto nivel a INH por alteración del gen katG permanecen sensibles a la ETH. Presenta resistencia cruzada con la tiacetazona.
La CIM de ETH para M. tuberculosis oscila entre 0,6 y 2,5μg/ml, y se alcanzan valores séricos de 20μg/ml a las 3–4h de una dosis oral de 0,5 a 1g. Pueden existir efectos adversos que incluyen intolerancia digestiva, hepatitis tóxica y diversos tipos de neurotoxicidad.
Amicacina, kanamicina, capreomicina y viomicinaLa amicacina y la kanamicina, junto con la STR, son aminoglucósidos similares estructuralmente y poseen actividad antituberculosa. La tobramicina y la gentamicina, por el contrario, no son efectivas en el tratamiento de la tuberculosis en las dosis habituales. La capreomicina y la viomicina son antimicrobianos peptídicos básicos que comparten el mecanismo de acción con los aminoglucósidos. La proporción de mutantes resistentes espontáneas es elevada, y oscila entre 10−3 y 10−5. No se absorben por vía oral y son de administración intramuscular.
No existe resistencia cruzada entre la STR y la capreomicina37, por lo que la capreomicina es activa frente a la mayoría de cepas resistentes a la STR. La CIM de la capreomicina en cepas sensibles oscila alrededor de los 10μg/ml. Suelen alcanzarse picos séricos de 30μg/ml. La capreomicina es menos tóxica que la viomicina, la amicacina y, especialmente, que la kanamicina, por lo que se la considera una alternativa para el tratamiento de la tuberculosis resistente. Algunas cepas resistentes a la capreomicina presentan resistencia cruzada con la amicacina y la kanamicina.
La amicacina es el antibiótico más potente del grupo, tanto in vitro como en el modelo animal, con CIM frente a M. tuberculosis de 1μg/ml. Se han obtenido concentraciones séricas de 10–30μg/ml después de la administración de una dosis intramuscular. Existe poca experiencia en el tratamiento de la tuberculosis por su precio elevado y mayor toxicidad. No obstante, puesto que suele ser más asequible la determinación de concentraciones séricas de amicacina que de STR o capreomicina, la amicacina puede ser una alternativa en aquellos enfermos con insuficiencia renal o en ancianos con alteraciones auditivas. Como es bien conocido, todos los antimicrobianos del grupo tienen efectos ototóxicos (tanto vestibulares como cocleares), así como nefrotóxicos.
TiosemicarbazonaEs uno de los fármacos antituberculosos menos potentes y más tóxicos. Su mecanismo de acción se desconoce. Es activo frente a M. tuberculosis con una CIM promedio de 1μg/ml; se obtienen picos séricos de 1 a 4μg/ml tras 1–2h de una dosis oral de 150mg.
La proporción de mutantes resistentes espontánea es elevada. Se utiliza, por su bajo coste, en algunos países en vías de desarrollo. No se dispone del fármaco ni en Europa ni en Estados Unidos. Entre otras reacciones adversas, puede producir reacciones cutáneas graves, hepatitis, anemia hemolítica, granulocitopenia y trastornos del equilibrio. En enfermos infectados por VIH se ha asociado el tratamiento de la tuberculosis con tiosemicarbazona a la aparición del síndrome de Stevens-Johnson y a necrólisis epidérmica grave38, por lo que se desaconseja su uso en este tipo de pacientes.
Rifamicinas: rifabutina, rifapentina y rifalazilLa RIF ha sido modificada para dar lugar a diferentes rifamicinas, entre las que se encuentran rifabutina, rifapentina y rifalazil. El mecanismo de acción y los mecanismos de resistencia son idénticos a los de la RIF con la que existe, generalmente, resistencia cruzada. Así, la resistencia de alto nivel debida a mutaciones en las posiciones 513-533 del gen rpoB suelen afectar a todas las rifamicinas, mientras que cepas con otras mutaciones del mismo gen, responsables de resistencia de bajo nivel a la RIF, pueden mantener la sensibilidad, particularmente a la rifapentina y al rifalazil25,39–41.
Todas ellas poseen, en función de las CIM, una mayor actividad in vitro que la RIF frente a M. tuberculosis, y también son mas activas en el modelo múrido de tuberculosis39. Por otra parte, su mayor vida media permite su dosificación una o 2 veces por semana. La rifabutina se absorbe bien por vía oral y alcanza concentraciones séricas de 0,5μg/ml después de 4h de una dosis de 300mg. Los efectos adversos son similares a los de la RIF e incluyen las interacciones con fármacos antirretrovirales. A pesar de esto, en los enfermos infectados por VIH tratados con inhibidores de las proteasas se recomienda tratar con 150mg de rifabutina en lugar de 600mg de RIF42. También se debería utilizar en los pacientes con tratamiento con ciclosporina o tacrolimus en los que se desaconseja la RIF. La rifapentina alcanza concentraciones séricas de 15μg/ml a las 5 o 6h de una dosis de 600mg. Cuando se ha comparado el tratamiento con rifapentina 2 veces a la semana con el tratamiento diario con RIF, los resultados han sido similares, aunque el número de recaídas en el grupo de la rifapentina ha sido ligeramente superior. No se dispone de rifapentina en España. En los ensayos en fase ii del rifalazil se ha detectado toxicidad que cuestiona su utilidad como fármaco antituberculoso39.
FluoroquinolonasEl núcleo central de la estructura de las moléculas de quinolona es un anillo 4-oxo-1,4-dihidroquinoleína que, en las nuevas quinolonas, incorpora en el carbono 6 un átomo de flúor (fluoroquinolonas). La actividad de las quinolonas tiene lugar en la ADN-girasa y la topoisomerasa iv. El desarrollo de la resistencia a las quinolonas en M. tuberculosis se debe a mutaciones que afectan la ADN-girasa o a proteínas de membrana que regulan la permeabilidad y el eflujo del fármaco. Se han descrito mutaciones en gyrA en gyrB43, así como una bomba de eflujo (gen lfra) que confiere una resistencia de bajo nivel44.
La actividad de las nuevas quinolonas sobre M. tuberculosis, su buena distribución tisular y celular, así como los escasos efectos adversos hacen que las fluoroquinolonas se hayan utilizado en la última década para el tratamiento de cepas MDR45, el tratamiento empírico en comunidades con una alta tasa de multirresistencia o el tratamiento de pacientes con reacciones adversas a los fármacos de primera línea. Recientemente se está investigando el uso de fluoroquinolonas para acortar el tratamiento de la tuberculosis sensible. La eficacia de las fluoroquinolonas se ha demostrado tanto en el modelo murino como en el de cultivo de macrófagos46–48. La CIM de ciprofloxacino frente a M. tuberculosis oscila entre 0,25 y 3μg/ml y la de ofloxacino entre 0,5 y 2,5μg/ml; se ha sugerido 2μg/ml como punto de corte para la sensibilidad49. Se obtienen picos séricos de 2,4 y 4,3μg/ml tras 1–2h de una dosis oral de ciprofloxacino de 500 o 750mg, respectivamente. Para ofloxacino, la concentración sérica después de 1 h de una dosis oral de 400mg es de 2,9μg/ml. Hay que tener en cuenta que la concentración en el tejido pulmonar de ciprofloxacino y ofloxacino es de aproximadamente 4 veces la observada en el suero. Moxifloxacino y gatifloxacino tienen CIM mucho más bajas que las demás fluoroquinolonas. Están en curso diversos ensayos que pretenden demostrar su eficacia in vivo. Se han descrito alteraciones de la glucemia con el uso de gatifloxacino. Una de las limitaciones del tratamiento con quinolonas es el desarrollo de resistencias, particularmente en tratamientos de cepas MDR en los que las fluoroquinolonas sean el único fármaco activo49. De hecho, el 51% de las cepas MDR en Filipinas son resistentes a las fluoroquinolonas50.
Nuevos fármacos antituberculosos en desarrollo39,51,52Los principales desafíos en el tratamiento de la tuberculosis son, por una parte, las cepas MDR (y XDR, virtualmente intratables) y, por otra, conseguir acortar la duración y la dosificación del tratamiento para facilitar la adherencia a éste. Para esto, se precisan fármacos que actúen rápidamente, que sean activos frente a las cepas resistentes y que sean capaces de eliminar las poblaciones “durmientes”. En los últimos años se han desarrollado análogos de los fármacos existentes que permitirían reducir la frecuencia de la dosificación (rifamicinas) o la duración del tratamiento (moxifloxacino, gatifloxacino). En cualquier caso, son necesarios nuevos fármacos de primera línea con mecanismos de acción diferentes (sin resistencias cruzadas con los demás antituberculosos), y que tengan el potencial de reducir la duración y la frecuencia de dosificación del tratamiento. A continuación se señalan algunas moléculas que están en desarrollo.
Diaril quinoleínas: TMC207De las diaril quinoleínas con actividad in vitro frente a M. tuberculosis, TMC207 es la más potente in vivo. Tiene actividad frente a las cepas sensibles, MDR y XDR de M. tuberculosis; no presenta resistencia cruzada con los demás antituberculosos, dado su mecanismo de acción sobre la bomba de protones de la adenosin trifosfato (ATP) sintetasa unida a la membrana53. Este mecanismo de acción es prometedor dada la poca similitud entre las proteínas humana y micobacteriana. Se han descrito cepas resistentes con mutaciones del gen atpE. Se absorbe bien por vía oral, su vida media es larga y permitiría una dosificación semanal. En el modelo múrido, TMC207 tiene una potente acción esterilizante.
Nitroimidazoles: PA-824, OPC-67683PA-824 es activo in vitro frente a cepas sensibles y resistentes a M. tuberculosis54. Aunque su mecanismo de acción no es totalmente conocido, parece actuar tanto en la síntesis proteica como de los lípidos de la pared55. Es un profármaco que requiere la reducción del grupo aromático por una glucosa-6-fosfato deshidrogenasa específica. Estudios farmacocinéticos en el modelo murino muestran una excelente penetración en los tejidos con una dosis bactericida mínima de 100mg/kg/día. OPC-67683 tiene una potente actividad in vitro e in vivo frente a M. tuberculosis. En el modelo múrido de tuberculosis, muestra una eficacia superior a la de los fármacos actualmente utilizados en el tratamiento. Sobre la base de la semejanza estructural y la existencia de resistencia cruzada es posible que el mecanismo de acción sea parecido, sino idéntico, al de PA-824. Tanto PA-824 como OPC-67683 están desprovistos de actividad mutagénica.
Diamina: SQ109Desarrollado originalmente en un intento de mejorar la molécula de ETB, SQ109 presenta diferencias estructurales y de diana, por lo que aparentemente constituye un nuevo compuesto antimicobacteriano. Presenta una excelente actividad in vitro frente a cepas sensibles y resistentes a INH, a RIF o a ETB56. El mecanismo de acción no se conoce con exactitud, aunque se cree que interfiere en la síntesis de la pared celular de una manera distinta a la del ETB. Ensayos clínicos en fase ia no encontraron efectos secundarios destacables y remarcaron una excelente distribución en los tejidos después de su administración oral57. La vida media es larga, lo que parece demostrar que una dosificación semanal sería suficiente.

