






La distrofia muscular de Duchenne (DMD) es la miopatía más frecuente en niños, con una prevalencia mundial de aproximadamente 0,5 por cada 10.000 varones. Se caracteriza por una debilidad muscular progresiva al inicio de la infancia con aparición posterior de complicaciones musculoesqueléticas, respiratorias y cardíacas que ocasionan discapacidad, dependencia y muerte prematura. Actualmente su tratamiento se fundamenta en medidas sintomáticas multidisciplinares que han modificado favorablemente el curso de la enfermedad, por lo que resulta crucial establecer unas directrices claras y actualizadas que permitan tanto una detección temprana de la enfermedad como un adecuado tratamiento y seguimiento de sus posibles complicaciones.
DesarrolloCon el fin de obtener una visión general de los aspectos abordados por las guías actuales y detectar aquellos en los que todavía no existe un consenso y su abordaje sea relevante, se realizó una revisión de la literatura en la base de datos biomédicas de los últimos 10años. El grado de evidencia y el nivel de recomendación de la información obtenida se clasificaron y ordenaron de acuerdo con los criterios de la American Academy of Neurology (AAN).
ConclusionesEl abordaje de la DMD debe ser multidisciplinar y ajustado al perfil del paciente y su grado de evolución clínica, comprendiendo, además del tratamiento basado en corticoides, medidas a nivel gastrointestinal, respiratorio, cardiaco, fisioterapéutico y ortopédico dirigidas a mejorar la calidad de vida de los pacientes. Los estudios genéticos desempeñan un papel clave en el manejo de la enfermedad, tanto en la detección de casos y posibles portadoras como en la caracterización de la mutación implicada y el desarrollo de nuevas terapias.
Duchenne muscular dystrophy (DMD) is the most common myopathy in children, with a worldwide prevalence of approximately 0.5 cases per 10,000 male births. It is characterised by a progressive muscular weakness manifesting in early childhood, with the subsequent appearance of musculoskeletal, respiratory, and cardiac complications, causing disability, dependence, and premature death. Currently, DMD is mainly managed with multidisciplinary symptomatic treatment, with favourable results in terms of the progression of the disease. It is therefore crucial to establish clear, up-to-date guidelines enabling early detection, appropriate treatment, and monitoring of possible complications.
DevelopmentWe performed a literature search of the main biomedical databases for articles published in the last 10years in order to obtain an overview of the issues addressed by current guidelines and to identify relevant issues for which no consensus has yet been established. The degree of evidence and level of recommendation of the information obtained were classified and ordered according to the criteria of the American Academy of Neurology.
ConclusionsDMD management should be multidisciplinary and adapted to the patient's profile and the stage of clinical progression. In addition to corticotherapy, treatment targeting gastrointestinal, respiratory, cardiac, and orthopaedic problems, as well as physiotherapy, should be provided with a view to improving patients’ quality of life. Genetic studies play a key role in the management of the disease, both in detecting cases and potential carriers and in characterising the mutation involved and developing new therapies.
La distrofia muscular de Duchenne (DMD) es una enfermedad de herencia recesiva ligada al cromosomaX que afecta a uno de cada 3.800-6.300 varones nacidos vivos1. Su prevalencia a nivel mundial es de aproximadamente 0,5 por cada 10.000 varones2, lo que equivale a 1.000 casos en España y 12.500 en la Unión Europea, convirtiéndose en la distrofia muscular más frecuente en niños. Se caracteriza por una debilidad muscular progresiva al inicio de la infancia y la aparición posterior de complicaciones que ocasionan discapacidad, dependencia y muerte prematura.
La mayoría de los pacientes se diagnostican entre los 3 y los 5años de edad3. La debilidad muscular sigue un curso progresivo hasta hacerse necesario el uso de una silla de ruedas antes de la adolescencia, mientras que simultáneamente o con posterioridad van surgiendo complicaciones respiratorias, ortopédicas y cardiacas4. También es frecuente la presentación de disfunciones neurocognitivas que, aunque no son progresivas5, suponen un impacto en el aprendizaje y en la calidad de vida de los pacientes. En los últimos años el adecuado manejo de las complicaciones6,7, junto con el uso de corticoides8, han permitido prolongar la supervivencia hasta la tercera o cuarta década de la vida9-13.
La DMD está causada por diversas mutaciones en el gen de la distrofina (DMD; locus Xp21.2)14 que conducen a la ausencia de una proteína subsarcolémica esencial para la estabilidad estructural del músculo y desencadena una grave degeneración muscular progresiva15. Existen variantes alélicas más leves, como la distrofia muscular de Becker (DMB), en la que la distrofina solo está disminuida y se mantiene la deambulación después de los 16años, formas intermedias entre DMD y DMB, y otras variantes que presentan afectación exclusivamente cardíaca o que cursan de forma asintomática o paucisintomática.
El manejo terapéutico de la DMD es fundamentalmente sintomático, basado en un conjunto de medidas protocolizadas cuyo objetivo es mejorar la funcionalidad y la calidad de vida de los pacientes, retrasar y tratar las complicaciones y prolongar la supervivencia. Para ello resulta de vital importancia establecer unas directrices claras que permitan tanto la detección temprana de la enfermedad como su adecuado seguimiento.
ObjetivosA pesar de que existen guías clínicas de consenso que abordan diferentes aspectos del manejo de la DMD8,16-18, en España todavía no se dispone de una guía actualizada que incorpore los avances más recientes de la enfermedad. El objetivo principal del presente documento es servir de guía a los profesionales sanitarios en el diagnóstico y tratamiento de la enfermedad y ayudarles en la toma de decisiones en todos los aspectos relevantes que conciernen al manejo de la enfermedad.
PoblaciónDado que la DMD presenta un patrón de herencia recesiva ligada al cromosomaX, suele afectar de forma mayoritaria a varones. No obstante, un 10% de las mujeres portadoras de la mutación muestran alguna de las manifestaciones propias de la enfermedad, en general con una expresión más leve, y que puede incluir o incluso afectar exclusivamente a la función cognitiva y/o cardiaca. Salvo una pequeña proporción de portadoras sintomáticas relacionada con reordenamientos cromosómicos, la mayoría deben su enfermedad a una inactivación no aleatoria del cromosomaX19-21. Este documento servirá de guía para mejorar la calidad en la atención de los pacientes diagnosticados con DMD, independientemente del sexo.
MetodologíaPara la elaboración de este documento se constituyó un grupo de trabajo formado por neurólogos, neuropediatras y rehabilitadores con experiencia en el tratamiento de pacientes con DMD pertenecientes a distintos centros hospitalarios españoles. Se realizó una revisión de la literatura en bases de datos (Medline, Pubmed y Cochrane) empleando la estrategia de búsqueda ((guidelines [MeSH Terms]) OR consensus document [MeSH Terms]) AND Duchenne (guidelines [MeSH Terms]) AND Duchenne y limitando los resultados a los últimos 10años. Adicionalmente, para España se utilizó Google Scholar. El grupo de trabajo clasificó y ordenó el grado de evidencia y el nivel de recomendación de la información recabada de acuerdo con los criterios de la American Academy of Neurology (AAN) (tabla 1)22.
Criterios de evaluación del grado de evidencia y el nivel de recomendación según la AAN
| Grados de evidencia | |
|---|---|
| Clase I | Ensayo clínico controlado y aleatorizado que reúne además las siguientes características: ciego, con objetivo primario claramente definido, con criterios de inclusión y exclusión claramente definidos y con registro adecuado de abandonos |
| Clase II | Ensayo clínico controlado y aleatorizado que no reúne al menos una de las características antes mencionadas |
| Clase III | Cualquier otro estudio controlado |
| Clase IV | Resto de estudios incluido consensos y opinión de expertos |
| Nivel de recomendación | |
|---|---|
| A | Establecido como efectivo, inefectivo o perjudicial (al menos 2 estudios clase I) |
| B | Probablemente efectivo, inefectivo o perjudicial (un estudio clase I o 2 estudios clase II) |
| C | Posiblemente efectivo, inefectivo o perjudicial (un estudio clase II o 2 estudios clase III) |
| U | Datos insuficientes o conflictivos |
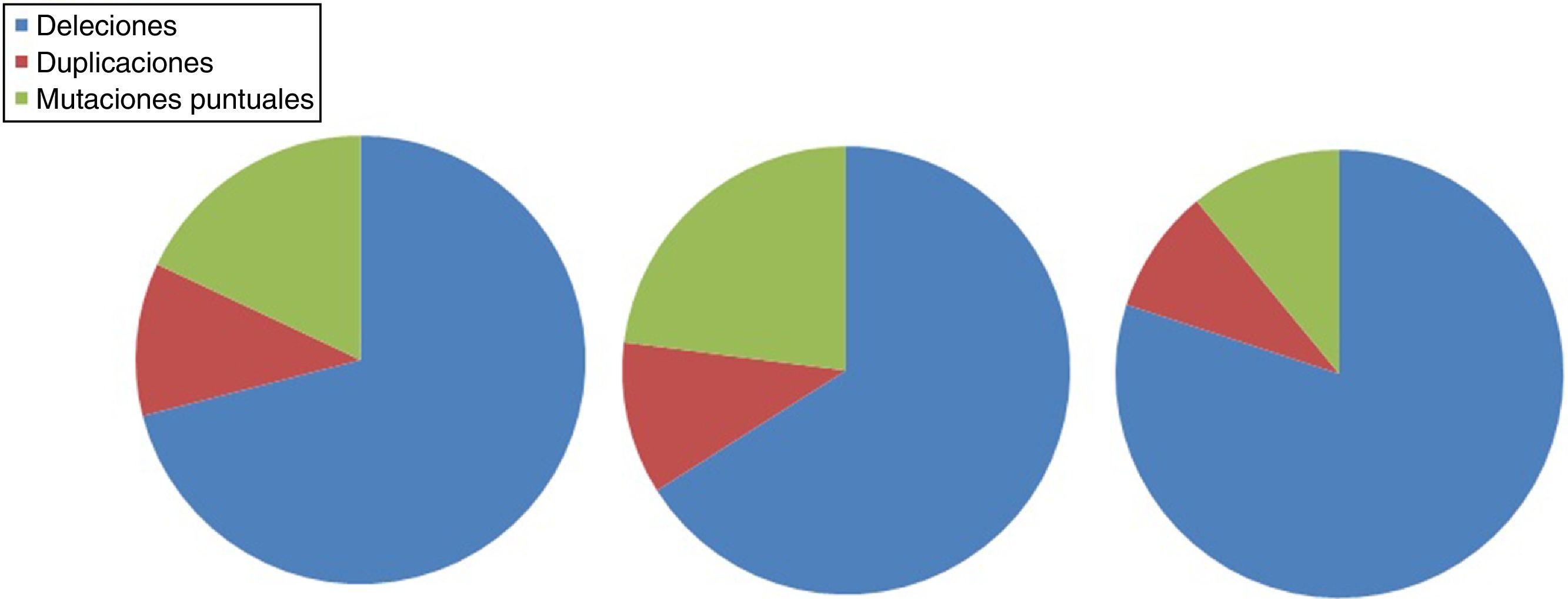
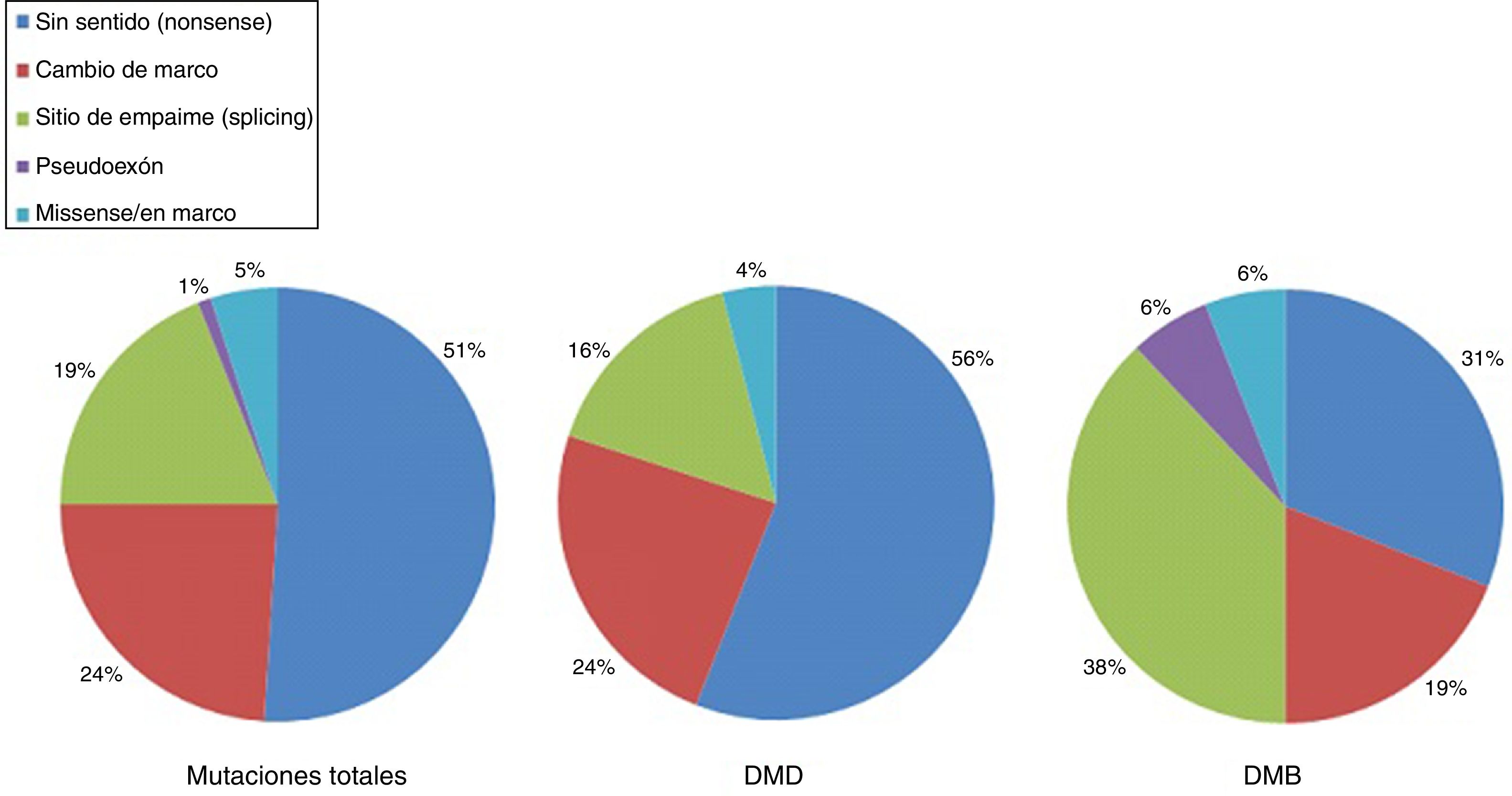
La DMD está causada por mutaciones en el gen de la distrofina, en el cromosomaX. La mayoría de pacientes presentan grandes reordenamientos génicos causados por deleción en uno o más exones (aproximadamente el 60-65%) o duplicación de exones (entre un 5-15%). En torno al 20% de casos presentan pequeñas mutaciones, bien sean puntuales causadas por el cambio de una base o la deleción o inserción de uno o varios nucleótidos23. Entre estas últimas, las más frecuentes son las mutaciones sin sentido (nonsense), que representan un 10% del total de los pacientes24,25. Las figuras 1 y 2 ilustran la frecuencia relativa de mutaciones en una serie extensa de distrofinopatías en pacientes españoles. Independientemente del tipo de mutación presente, salvo en las mutaciones puntuales, la alteración genética tiene que romper el marco de lectura del ARNm que sintetiza la distrofina, lo que conduce a la producción de una proteína no funcional26. Sin embargo, en el 10% de los casos esta regla no se cumple27,28 debido a la existencia de un reordenamiento secundario oculto, o salto espontáneo de la lectura de un exón, que restablecería el marco. En el caso de mutaciones puntuales los mecanismos patogénicos son variados, siendo el más frecuente el de las mutaciones sin sentido que generan un codón de parada prematura de la lectura ribosomal sin ruptura del marco de lectura, lo que produce una distrofina aberrante no funcional que igualmente ocasiona un fenotipo de DMD29.

Distribución de las mutaciones genéticas asociadas a DMD y BMD en población española. Adaptado de Juan-Mateu et al.25.

Distribución de las mutaciones puntuales asociadas a DMD y BMD en población española. Adaptado de Juan-Mateu et al.25.
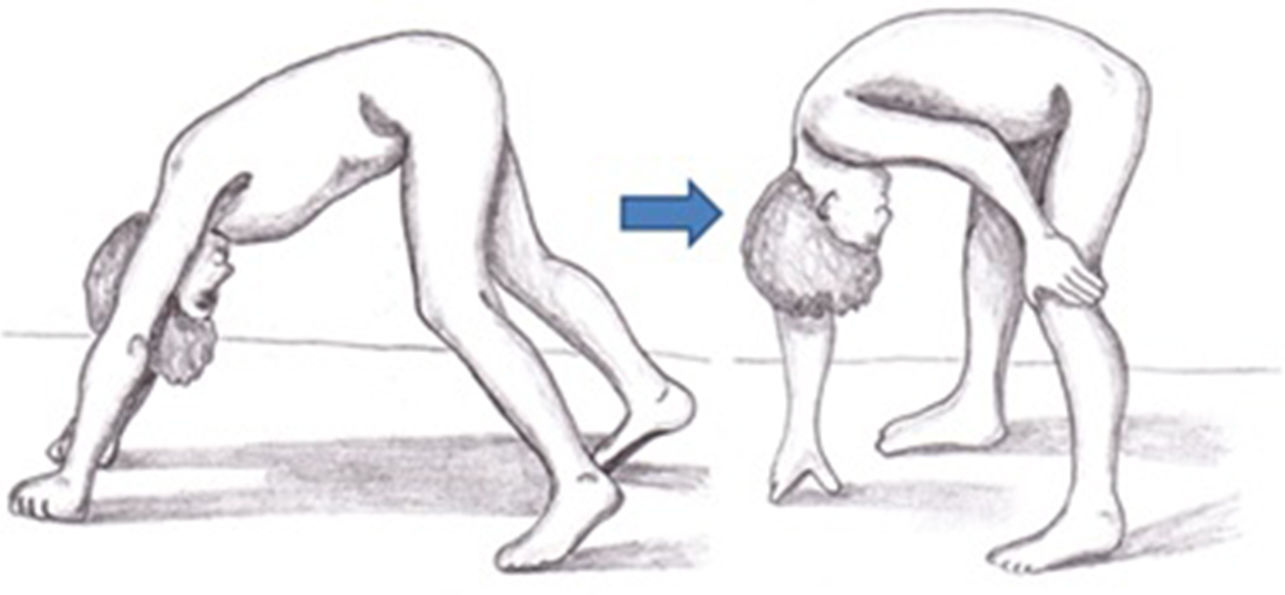
En pacientes con DMD los síntomas no son visibles al nacimiento, pero una proporción de ellos manifiesta tempranamente un retraso en el neurodesarrollo. Los síntomas iniciales suelen manifestarse durante los tres primeros años de vida con dificultades en la marcha, caídas frecuentes, dificultad para subir escaleras y levantarse del suelo o tendencia a caminar de puntillas (tabla 2)30. La exploración neurológica en esta primera etapa puede mostrar debilidad axial, signo de Gowers (fig. 3), pseudohipertrofia muscular o leve retracción aquílea26. Igualmente es en esta etapa cuando comienzan a detectarse, en algunos casos, los problemas cognitivos. Los niños progresan favorablemente en el desarrollo de sus habilidades motoras hasta los 4-6años, a un ritmo más lento, alcanzando una fase de meseta entre los 4 y los 8años31, seguida de un declive progresivo en la que la pérdida de fuerza muscular se acentúa y las capacidades adquiridas se van perdiendo8,32. En esta fase se desarrollan contracturas y retracciones en las articulaciones menos movilizadas26, impactando de manera negativa en su confort y en su funcionalidad a nivel motor33. La reducción de la actividad física, sumada al tratamiento con corticoides, favorece el desarrollo de sobrepeso, que incide sobre la movilidad, fomenta el desarrollo de osteoporosis e implica un riesgo de fracturas elevado34. Hasta hace unos años los pacientes necesitaban silla de ruedas antes de la adolescencia26. Sin embargo, gracias a la mejora de las medidas de soporte y la terapia con corticoides se ha retrasado la pérdida de ambulación8.
Resumen de las principales manifestaciones clínicas en las diferentes fases de la DMD
| Ambulatoria temprana | Ambulatoria tardía | No ambulatoria temprana | No ambulatoria tardía |
|---|---|---|---|
| Debilidad en miembros inferioresManiobra de GowersMarcha con balanceo de caderas.Marcha de puntillasLimitación para subir escalerasImposibilidad para saltarDificultad en aprendizaje y problemas de conducta | Marcha cada vez más dificultosaPérdida de la habilidad para subir escaleras y levantarse del sueloPrimeros síntomas de escoliosis | Pérdida de la marchaCapacidad para mantenerse de pieDesarrollo de escoliosis | Debilidad progresiva en extremidades superiores e incapacidad para mantenerse sentadoComplicaciones cardiacas y respiratorias |

La afectación de isoformas de la distrofina que se expresan selectivamente en otros órganos como el cerebro26 explica que aproximadamente el 20-34% de los pacientes presenten un déficit intelectual, síntomas del espectro autista u otras alteraciones cognitivo-conductuales35,36.
Fase no ambulatoria temprana - no ambulatoria tardíaEn la mayoría de individuos con DMD la pérdida de la marcha ocurre entre los 12-14años. Esto facilita la aparición de complicaciones ortopédicas graves, como la escoliosis, que afecta al 90% de los pacientes como consecuencia de la debilidad de los músculos paraespinales, a la vez que surgen complicaciones respiratorias y cardíacas. Los problemas respiratorios se inician durante el sueño con hipoventilación nocturna, provocando apneas periódicas, cefaleas matutinas, náuseas, fatiga, pérdida de apetito y deterioro cognitivo37,38. La alteración de la función respiratoria, agravada por la escoliosis, ha supuesto durante años la principal causa de muerte. Sin embargo, el adecuado manejo respiratorio y diferentes formas de soporte ventilatorio están condicionando un aumento de los problemas cardiacos (miocardiopatías y arritmias) como causa importante de fallecimiento en estos pacientes.
Las principales características clínicas de los pacientes de DMD a lo largo de las distintas fases de la enfermedad se sintetizan en la tabla 2.
Conviene señalar que existe variabilidad individual en el ritmo de progresión y gravedad de la enfermedad. Incluso hermanos con idéntica mutación en el gen de la distrofina han mostrado diferencias clínicas relevantes39, lo que sugiere la existencia de factores modificadores (ambientales o genéticos) que condicionan el fenotipo individual.
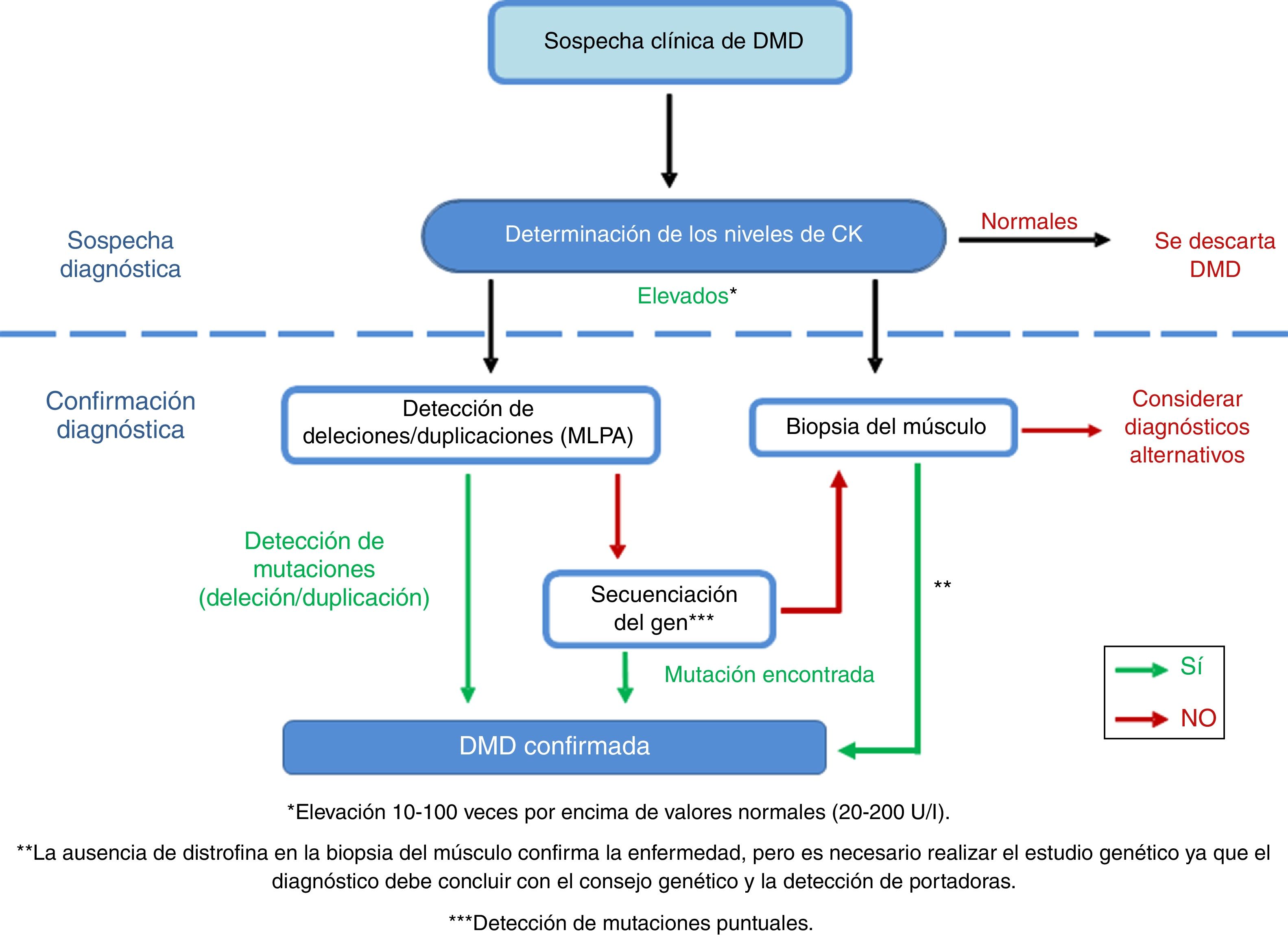
DiagnósticoEl diagnóstico de la patología debe ser lo más temprano posible, rápido y preciso, a fin de garantizar un inicio precoz de las intervenciones sobre el paciente. Ante la sospecha clínica de DMD se deben determinar los niveles séricos de creatina quinasa (CK; fig. 4)40 [IV, U], que en los pacientes con DMD superan 10-100 veces26 los valores normales41. Si ya existen antecedentes familiares se debe realizar una determinación de los niveles de CK ante la más mínima sospecha [IV, U]8,26. El 10% de las mujeres portadoras también presentan una elevación de los niveles de CK21. Como consecuencia de la destrucción muscular se produce en paralelo una elevación de transaminasas que, de forma errónea, puede conducir a la sospecha de enfermedad hepática en niños con DMD presintomáticos.

Adaptación del algoritmo diagnóstico de DMD, desde la sospecha del mismo hasta su confirmación.
Fuente: Camacho26.
Cuando la sospecha de DMD sea alta, se recomienda comenzar con un estudio genético que en muchos casos puede evitar la biopsia muscular, que es una prueba de diagnóstico invasiva26. Se suele comenzar empleando la Multiplex Ligation-dependent Probe Amplification (MLPA) para detectar los exones implicados en las deleciones y duplicaciones42 [III, C]. Si los resultados de la prueba son positivos y el paciente presenta un fenotipo compatible, ya se puede establecer el diagnóstico de DMD. Si los resultados son negativos, debe secuenciarse el gen [III, C] para buscar mutaciones puntuales o pequeñas deleciones/duplicaciones43,44. Resulta necesario caracterizar por completo la mutación para evaluar su influencia en el estado del marco de lectura de la proteína, principal determinante de la diversidad fenotípica de las distrofinopatías45-47.
Si el estudio genético no identifica ninguna mutación pero las concentraciones de CK se encuentran aumentadas y están presentes los signos o síntomas compatibles con la enfermedad, se debe llevar a cabo una biopsia del músculo para confirmar o descartar el diagnóstico8,48 [IV, U].
El complemento a las pruebas genéticas descritas comprende la realización de una biopsia muscular. El estudio con microscopia óptica muestra un patrón distrófico con desestructuración de la arquitectura fascicular del músculo, necrosis y regeneración de fibras musculares, e incremento del tejido conectivo-adiposo endomisial. Mediante técnicas de inmunohistoquímica se comprueba la deficiencia de distrofina49 [III, C].
Si se opta en primera instancia por realizar una biopsia del músculo y se diagnostica la enfermedad comprobando el déficit de distrofina, a continuación es obligatorio realizar las pruebas genéticas para identificar el tipo de mutación causal26 [IV, U]. Es muy recomendable que, en caso de hacer la biopsia, se realice el cultivo de mioblastos con fines de investigación [IV, U].
Tratamiento y seguimiento de pacientesLa historia natural de la DMD ha evolucionado en los últimos años gracias a la instauración de un tratamiento multidisciplinar precoz que incluye la administración de corticoides y un adecuado manejo y seguimiento respiratorio, cardiaco, nutricional, fisioterapéutico y ortopédico, estabilizando o disminuyendo el ritmo de progresión de la enfermedad.
En la presente guía se presenta una síntesis actualizada de los distintos abordajes en cuanto al manejo y tratamiento de la DMD y sus complicaciones, contemplando tanto las estrategias tradicionales como las terapias más novedosas, que se resumen de forma esquemática en el Anexo 1.

El manejo tradicional se basa en el uso de corticoides gracias a sus efectos beneficiosos a largo plazo sobre la función motora, cardiaca y respiratoria (Anexo 1)50 [I, A]. Se recomienda comenzar el tratamiento cuando la función motriz del niño alcanza un nivel estable (4-6años de edad) [IV, U]. El uso de escalas clínicas y funcionales puede ayudar a reconocer el momento de estabilidad51,52 [III, C]. Existe controversia acerca de si el tratamiento con corticoides ha de mantenerse tras la pérdida de deambulación. Los estudios más recientes han mostrado preservar la fuerza de las extremidades superiores, reducir la progresión de la escoliosis y retrasar la afectación cardiaca y pulmonar53 [IV, U]. El programa nacional de vacunación recomendado debe estar completo antes de iniciar el tratamiento con corticoides y debe quedar manifiesta la inmunidad a la varicela8,54 [IV, U]. Los corticoides utilizados son prednisona y deflazacort. La administración discontinua es menos eficaz pero se asocia a menos efectos adversos55. Los regímenes más utilizados contemplan la administración de: a)0,75mg/kg/día de prednisona o b)0,9mg/kg/día de deflazacort, y c)0,75mg/kg/día de prednisona a días alternos o bien durante 10días, seguidos de 10días de descanso. Aunque ambos principios activos han demostrado una eficacia similar mejorando la fuerza y la función muscular50 [I, A], los trabajos más recientes indican que el tratamiento continuado con deflazacort es, en general, más seguro56 [I, A].
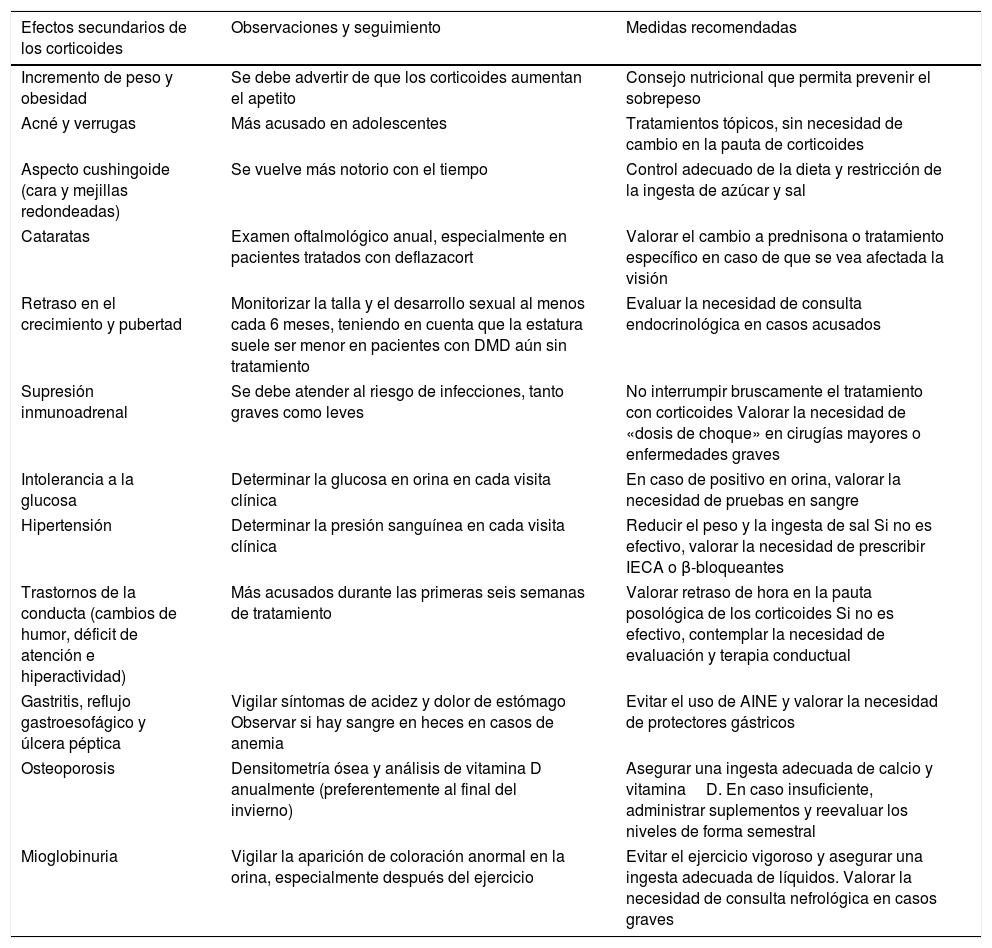
La prednisona y la prednisolona tienen efecto antiinflamatorio y aumentan la fuerza del músculo esquelético57, mientras que deflazacort actúa además favoreciendo la regeneración muscular a nivel esquelético58,59. Una proporción importante de pacientes no toleran el uso crónico de corticoides por la aparición de efectos adversos (tabla 3), mientras que otros no responden de forma satisfactoria al tratamiento60. Generalmente la dosis de corticoides se incrementa a medida que el niño crece, siempre que los efectos secundarios sean manejables, hasta que alcanza aproximadamente los 40kg16. Si no se producen efectos adversos relevantes, la dosis establecida se mantiene durante la fase no ambulatoria. Si los efectos secundarios son difícilmente manejables, la dosis del fármaco se reducirá un 30% antes de plantear su retirada.26.
Principales efectos adversos del tratamiento con corticoides, seguimiento y medidas terapéuticas recomendadas
| Efectos secundarios de los corticoides | Observaciones y seguimiento | Medidas recomendadas |
|---|---|---|
| Incremento de peso y obesidad | Se debe advertir de que los corticoides aumentan el apetito | Consejo nutricional que permita prevenir el sobrepeso |
| Acné y verrugas | Más acusado en adolescentes | Tratamientos tópicos, sin necesidad de cambio en la pauta de corticoides |
| Aspecto cushingoide (cara y mejillas redondeadas) | Se vuelve más notorio con el tiempo | Control adecuado de la dieta y restricción de la ingesta de azúcar y sal |
| Cataratas | Examen oftalmológico anual, especialmente en pacientes tratados con deflazacort | Valorar el cambio a prednisona o tratamiento específico en caso de que se vea afectada la visión |
| Retraso en el crecimiento y pubertad | Monitorizar la talla y el desarrollo sexual al menos cada 6 meses, teniendo en cuenta que la estatura suele ser menor en pacientes con DMD aún sin tratamiento | Evaluar la necesidad de consulta endocrinológica en casos acusados |
| Supresión inmunoadrenal | Se debe atender al riesgo de infecciones, tanto graves como leves | No interrumpir bruscamente el tratamiento con corticoides Valorar la necesidad de «dosis de choque» en cirugías mayores o enfermedades graves |
| Intolerancia a la glucosa | Determinar la glucosa en orina en cada visita clínica | En caso de positivo en orina, valorar la necesidad de pruebas en sangre |
| Hipertensión | Determinar la presión sanguínea en cada visita clínica | Reducir el peso y la ingesta de sal Si no es efectivo, valorar la necesidad de prescribir IECA o β-bloqueantes |
| Trastornos de la conducta (cambios de humor, déficit de atención e hiperactividad) | Más acusados durante las primeras seis semanas de tratamiento | Valorar retraso de hora en la pauta posológica de los corticoides Si no es efectivo, contemplar la necesidad de evaluación y terapia conductual |
| Gastritis, reflujo gastroesofágico y úlcera péptica | Vigilar síntomas de acidez y dolor de estómago Observar si hay sangre en heces en casos de anemia | Evitar el uso de AINE y valorar la necesidad de protectores gástricos |
| Osteoporosis | Densitometría ósea y análisis de vitamina D anualmente (preferentemente al final del invierno) | Asegurar una ingesta adecuada de calcio y vitaminaD. En caso insuficiente, administrar suplementos y reevaluar los niveles de forma semestral |
| Mioglobinuria | Vigilar la aparición de coloración anormal en la orina, especialmente después del ejercicio | Evitar el ejercicio vigoroso y asegurar una ingesta adecuada de líquidos. Valorar la necesidad de consulta nefrológica en casos graves |
Ambos fármacos son los únicos agentes de terapia génica que han logrado superar el desarrollo preclínico y alcanzar la fase de ensayos clínicos aportando datos sobre eficacia y seguridad que han derivado en su aprobación como medicación por parte de las agencias reguladoras61.
Como se ha comentado previamente, existe un 10% de casos de DMD originados por mutaciones sin sentido (nonsense) que dan lugar a proteínas truncadas no funcionales.
Atalureno facilita la lectura ribosómica del codón de parada prematura del ARNm que aparece en las mutaciones sin sentido, lo que da lugar a la transformación de una proteína truncada no funcional en otra funcional62. Su eficacia y seguridad, determinadas a través de varios ensayos clínicos63-65 [II, B, III], han propiciado su autorización de forma condicional por la EMA en julio de 2014 para el tratamiento de la DMD debida a una mutación sin sentido en el gen de la distrofina, en pacientes ambulantes a partir de 5 o más años de edad62.
Eteplirsen es un oligonucleótido antisentido de síntesis diseñado para saltar el exón 51, permitiendo recuperar la pauta de lectura en algunas deleciones específicas de la región central del gen (13% de los casos de DMD). Por el momento ha mostrado mejorar los niveles de distrofina en un ensayo piloto [III, C], lo que ha llevado a su aprobación por la FDA.
Manejo musculoesqueléticoPromover el mantenimiento de una movilidad amplia y simétrica debe llevarse a cabo concertadamente por especialistas en patología neuromuscular, médicos rehabilitadores, fisioterapeutas y cirujanos ortopédicos16 [III, C]. Conviene además realizar una evaluación semestral para identificar los factores de riesgo e instaurar medidas y recomendaciones pertinentes [IV, U]. Dicha evaluación debe incluir pruebas como la determinación de la fuerza mediante la escala del Medical Research Council (MRC); la realización de pruebas funcionales cronometradas y el test de la marcha durante 6minutos66; también son recomendables las escalas de función motora, como la North Star Ambulatory Assessment en la fase ambulatoria67 o la Performance of Upper Limb Scale68 en la fase no ambulatoria [III, C]. Debe realizarse una medición de la amplitud de movimiento de las articulaciones. Adicionalmente, el uso de dispositivos externos ayuda a prevenir o minimizar las contracturas y/o deformidades, ya que están destinados al correcto mantenimiento postural69 [II]. Concretamente, las ortesis tobillo-pie de uso nocturno son adecuadas en todas las fases de la enfermedad70,71 [III, C], mientras que las ortesis rodilla-tobillo-pie diurnas, e incluso el uso de bipedestadores, resultan especialmente útiles en las últimas etapas ambulatorias y en el comienzo de las no ambulatorias71,72 [IV, U]. La natación es un deporte excelente para estos pacientes. Por el contrario, deben evitar el ejercicio de alta intensidad y de forma excéntrica26 [IV, U].
Con el objetivo de retrasar la pérdida de la marcha y evitar las complicaciones derivadas de la sedestación se han desarrollado programas de prolongación de la marcha con ortesis ligeras cuya implantación debe iniciarse en las últimas fases de deambulación73 antes de los 3meses del cese de la deambulación [III, C]. Las deformidades graves en pie equino deben corregirse mediante una tenotomía de Aquiles seguida de la colocación de la ortesis a los 2-3días y el inicio del reentrenamiento de la marcha74 [IV, U], especialmente en casos donde el grado de la retracción interfiere en el patrón de marcha o es de carácter asimétrico. En cuanto al desarrollo de escoliosis, debe monitorizarse a lo largo de todo el crecimiento. Una vez alcanzada la madurez esquelética se evaluará la necesidad de practicar una cirugía de fijación espinal de la columna para prevenir el empeoramiento y reducir la tasa de disminución de la capacidad respiratoria75 [III, C]. En la fase no ambulatoria resulta vital la figura del terapeuta ocupacional76 [IV, U].
Manejo respiratorioLas directrices para un correcto manejo respiratorio comprenden la incorporación de un neumólogo y un terapeuta especializado en fisioterapia respiratoria77-79 [IV, U]. Durante la fase ambulatoria, la capacidad vital forzada (CVF) ha de evaluarse anualmente. Tras la pérdida de la marcha es preciso estrechar el seguimiento cada 6meses26: la CVF en sedestación, la saturación de O2 con pulsioximetría y el flujo máximo de tos26 [IV, U]. Es importante realizar estudios de sueño cuando los valores de CVF son inferiores al 60% para detectar hipoventilación nocturna y síndromes de apneas-hipopneas obstructivas del sueño80 [III, C]. El control precoz de estas complicaciones es vital para prevenir complicaciones cardiacas y el deterioro de la función cognitiva81-83 [II, III; C]. Teniendo en cuenta el alto riesgo de infecciones respiratorias, debe incluirse una inmunización frente a patógenos como el neumococo (mayores de 2años) y la gripe (a partir de 6meses). No existe contraindicación en pacientes tratados con corticosteroides; sin embargo, estos pueden presentar una respuesta inmune disminuida. En caso de infección, con independencia del uso de antibióticos, es importante además el uso de tos asistida (fisioterapia respiratoria) para evitar complicaciones, especialmente en estados avanzados de la enfermedad84 [IV, U].
Manejo cardiacoLa afectación cardiaca es una complicación que toma protagonismo, convirtiéndose en una de las causas principales de mortalidad. Se caracteriza por una disfunción ventricular izquierda que evoluciona a una miocardiopatía dilatada que desemboca en insuficiencia cardíaca y arritmias85,86. A diferencia de otras miocardiopatías, suele cursar de forma asintomática dada la inmovilidad de los pacientes, por lo que su diagnóstico y su evaluación se basan en las pruebas pertinentes85,86. La evaluación debería incluir ECG y ecocardiograma al diagnóstico, seguida de revisiones bianuales que pasarían a anuales a partir de los 10años. La resonancia magnética cardiaca se está incorporando al seguimiento de los pacientes por su mayor sensibilidad en la detección de disfunción cardíaca y fibrosis87,88 [III, C].
La opción terapéutica de primera línea es el uso de inhibidores de la enzima convertidora de angiotensina (IECA)16,85. Estudios recientes apoyan su empleo previo a la aparición de signos de disfunción ventricular para tratar de retrasar su inicio89,90 [I, A]. En caso de intolerancia pueden emplearse los inhibidores del receptor de la angiotensina (IRA)91 [I, B]. Otro grupo que ha mostrado efecto cardioprotector son los antagonistas del receptor de aldosterona (ARA), aunque su uso como alternativa a los IECA o como terapia combinada permanece aún por definir92 [I, U]. El uso de β-bloqueantes es más controvertido, ya que no existe evidencia suficiente que acredite un claro beneficio en cardiopatías pediátricas de diferentes etiologías93 [II, C]; tampoco mostró ventajas cuando se administró asociado a un IECA92 [II, C].
El inicio de la terapia con agentes antiarrítmicos para controlar la fibrilación atrial y las arritmias ventriculares se instaura tras un fallo cardiaco o un episodio importante de arritmias85 [IV, U]. La aparición de hipertensión arterial sistémica debe ser tratada, al igual que las alteraciones del ritmo cardiaco8,16 [IV, U].
Manejo gastrointestinal y nutricionalDurante la enfermedad los pacientes con DMD presentan un gran riesgo de tener problemas de peso. Por ello es necesario un seguimiento nutricional periódico y ajustes en la dieta. En estadios avanzados de la enfermedad la disfagia94 puede ocasionar pérdida de peso, acentuando la pérdida gradual de la fuerza muscular respiratoria. En este caso se puede considerar la alimentación por sonda nasogástrica y botón gástrico posteriormente. Cuando se produce una pérdida de peso sin cambios en la ingesta se debe valorar la posibilidad de un mayor gasto energético debido al trabajo respiratorio para descartar la necesidad de instaurar ventilación no invasiva (VNI) o adaptar los parámetros de la misma16 [IV, U].
Los pacientes en fase no ambulatoria son susceptibles de sufrir reflujo gastroesofágico y esofagitis debido a la afectación de la musculatura esofágica. De forma general se trata con antagonistas del receptor H2 e inhibidores de la bomba de protones, solos o en combinación con procinéticos, sucralfato y antiácidos neutralizantes95 [III, C]. Los pacientes son propensos al estreñimiento, por lo que se debe asegurar la hidratación y la ingesta alimentaria adecuada. De forma puntual se pueden utilizar laxantes y enemas, y en casos persistentes leche de magnesio, lactulosa o polietilenglicol.
Manejo cognitivo y psicosocialLa DMD es una enfermedad eminentemente muscular, pero a la vez puede afectar al SNC con diversas manifestaciones, como retraso del neurodesarrollo en la infancia temprana96, problemas cognitivos y del aprendizaje en la etapa escolar51 y una elevación significativa de la incidencia de trastornos neuropsiquiátricos del espectro autista, déficit de atención e hiperactividad, conductas obsesivo-compulsivas y trastornos afectivos97-99, incluso a veces precediendo a los síntomas musculares100. En general, la incidencia de estos trastornos en diferentes cohortes de pacientes oscila entre el 20 y el 30%, adquiriendo la condición de grave en el 5% de los casos96. Estos problemas se deben evaluar de forma específica a través de escalas estandarizadas, como el cociente de desarrollo de Griffith101, la escala de Bayley-III51 o la escala de inteligencia de Weschler35, además de otros cuestionarios específicos [III, C]. En función del resultado, se indicará la necesidad de logopedia, psicoterapia o adaptación curricular a nivel escolar26 [IV, U]. Para muchos padres, el estrés causado por los problemas psicosociales del niño y las dificultades para conseguir su reconocimiento excede incluso al estrés relacionado con los aspectos físicos de la enfermedad, por lo que la búsqueda de apoyo psicológico puede resultar beneficiosa para ambos102.
Precauciones con la anestesiaSe recomienda el uso de anestésicos por vía intravenosa, dado el riesgo de hipertermia maligna y rabdomiólisis de los anestésicos inhalados, como halotano o isoflurano. Asimismo, los relajantes musculares despolarizantes están contraindicados26 [IV, U].
Asesoramiento genético familiarPara el asesoramiento genético es imprescindible detectar a las portadoras de una mutación. Pero hay que tener presente que un tercio de las madres de hijos afectados en realidad no lo son, por lo que su riesgo de transmisión es mínimo. Existe la posibilidad de un mosaicismo germinal que puede dar lugar a la existencia de mutaciones inesperadas en los óvulos de mujeres aparentemente no portadoras103, por lo que para una prevención segura habría de recomendarse estudio prenatal/preimplantacional en todas las madres.
La importancia del asesoramiento genético en la DMD reside, por un lado, en identificar a las posibles portadoras mediante las pruebas genéticas pertinentes. Por otro lado, estas pruebas aportarán la información clínica requerida para conocer la variación genética causante de la patología en cada caso y, de este modo, determinar la elegibilidad de los pacientes para ser incluidos en ensayos clínicos de valoración de futuras terapias para mutaciones específicas104.
Conclusiones e implementaciónLa DMD es una enfermedad grave que ocasiona una pérdida precoz de la deambulación y el desarrollo de complicaciones que conllevan una pérdida de la calidad de vida y una muerte prematura. A pesar de que en la actualidad no se dispone de un tratamiento curativo, existen estrategias que permiten retrasar la evolución natural de la enfermedad y la aparición de las complicaciones. Para poner en marcha dichas estrategias de forma efectiva son necesarios un diagnóstico preciso y temprano, así como la implantación de un plan de seguimiento predefinido y multidisciplinar. En cuanto al proceso diagnóstico, las recomendaciones en primera instancia ante la sospecha fundada de un caso de DMD comprenden la realización de un estudio genético para detectar posibles mutaciones en el gen de la distrofina, que ofrece un resultado inequívoco y, a la vez, no resulta invasivo26. Asimismo, su especial interés radica en la posibilidad de identificar la mutación específica causante de la patología que, a su vez, permitirá orientar futuras terapias dirigidas específicamente al gen mutado y determinar la elegibilidad de los pacientes para dichos estudios104.
El diagnóstico debe completarse con el estudio de portadoras. El actual manejo en la DMD consiste en el uso de corticosteroides. A pesar de existir problemas de tolerabilidad y refractariedad en algunos pacientes, en la mayoría han mostrado notables efectos beneficiosos sobre la función cardiaca, pulmonar y motora105, incluso tras la pérdida de la deambulación53. A este eje terapéutico fundamental han de añadirse medidas a nivel respiratorio, cardiaco, fisioterapéutico, ortopédico y gastrointestinal y nutricional, todas ellas dirigidas a reducir los síntomas y mejorar la calidad de vida de los pacientes, para lo que se requiere un equipo multidisciplinar especializado en el manejo de esta patología8. Los avances en cuanto al manejo respiratorio, especialmente la instauración de la ventilación nocturna, han aumentado significativamente la supervivencia en la DMD79,106.
El tratamiento de la miocardiopatía asociada a la DMD ha de instaurarse previamente a la aparición de signos de funcionamiento cardiaco anormal8 para garantizar mejoras en la evolución de la cardiomiopatía92. En lo que respecta al manejo musculoesquelético, resulta de vital importancia monitorizar el desarrollo de escoliosis, al mismo tiempo que se fomente una rehabilitación. Por tanto, la piedra angular en el cuidado óptimo de aquellos que sufren DMD es que la atención prestada debe ser de espíritu multidisciplinar, fomentando la colaboración entre profesionales sanitarios de diversas especialidades. En conclusión, es importante tener presente que, a pesar de los importantes avances logrados con el tratamiento basado en corticoides en cuanto a la prolongación de esperanza de vida de los pacientes, así como las últimas novedades en terapia génica como son atalureno y eterlipsen, la investigación en torno a la DMD debe continuar para abrir a la medicina actual una ventana a nuevos conceptos y estrategias que la aproximen a un cada vez más efectivo manejo de la enfermedad.
FinanciaciónEl soporte editorial fue proporcionado por Patricia Ortega, del Departamento Médico de Adknoma Health Research, S.L., y fue financiado por PTC Therapeutics S.L. mediante una beca educacional independiente. La elaboración, redacción y opiniones expresadas en este trabajo corresponden exclusivamente a los autores.
AutoríaTodos los autores proporcionaron su experiencia profesional en la concepción, diseño y realización del trabajo, así como en la búsqueda e interpretación de los datos, participando en la redacción del texto y sus revisiones, y aprobando su versión final.
Conflicto de interesesAna Camacho, Julita Medina, Juan Jesús Vilchez Padilla, Andrés Nascimento y Marcos Madruga declaran haber sido asesores de PTC Therapeutics y haber recibido remuneraciones por impartir charlas formativas. El resto de los autores declaran no tener ningún conflicto de intereses.

