





El pioderma gangrenoso (PG) fue descrito por primera vez en 1916 por Brocq, bajo el nombre de phagédénisme géométrique. En 1930 Brunsting et al. volvieron a describir la entidad y acuñaron el nombre actual, dado que era una úlcera necrótica y de supuesta etiología infecciosa.
Es una dermatosis inflamatoria primariamente estéril, ulcerativa e infrecuente, que se caracteriza por lesiones ulcerativas recurrentes, con unos hallazgos clínicos distintivos. Suele iniciarse como una pústula o nódulo doloroso, que rápidamente evoluciona a una úlcera de crecimiento rápido y progresivo, con bordes violáceos, eritema perilesional, fondo fibrinoso y exudado purulento o hemorrágico. No presenta características histológicas específicas, por lo que el diagnóstico de la entidad se basa en la exclusión de otras posibles causas de úlceras.
El PG puede presentarse en ausencia de patología asociada o en el contexto de alguna enfermedad sistémica.
La entidad se engloba dentro de las denominadas dermatosis neutrofílicas, entre las que, además del PG, se encuentran el síndrome de Sweet, la dermatosis neutrofílica del dorso de las manos, la enfermedad de Behçet, la vasculitis pustulosa, la enfermedad de Sneddon-Wilkinson, el eritema elevatum diutinum, la dermatosis IgA neutrofílica intraepidérmica, la urticaria neutrofílica y la hidradenitis ecrina neutrofílica. Todas ellas tienen en común su mecanismo patogénico, los hallazgos histológicos –suele observarse un infiltrado neutrofílico difuso y perivascular-, así como la ausencia de agente infeccioso identificable y la frecuente asociación a enfermedades internas.
EpidemiologíaAunque no hay datos epidemiológicos precisos, se estima que la incidencia de PG es de alrededor de 3-10 casos por millón de habitantes y año1,2. En la revisión de Suárez-Pérez et al. se establece una incidencia de 3,26 casos por millón de habitantes y año, en su población de referencia3. La máxima incidencia se sitúa entre los 20-50 años, con un leve predominio del sexo femenino, pero se han descrito casos en cualquier edad y se estima que los casos pediátricos representan un 4% del total. Algunos autores, además, describen una incidencia similar en ambos sexos1,3–5. Recientemente, Farhi et al. demostraron, en un estudio multivariante en 2.402 pacientes diagnosticados de enfermedad inflamatoria intestinal (EII), que el PG se asociaba significativa e independientemente a la raza negra de origen africano (p<0,003)6.
Etiología y patogeniaAunque se desconoce exactamente la patogenia y la etiología del PG, se cree que algunos mecanismos mediados inmunológicamente juegan un papel importante.
Inicialmente, en el momento de su descripción, Brunsting et al. creían que la causa era una infección bacteriana (estreptocócica o estafilocócica). Posteriormente, Fulbright et al.7 sugirieron que se pudiera tratar de una respuesta inmune aberrante a factores no identificados. La frecuente asociación a enfermedades sistémicas con una patogenia supuestamente autoinmune, junto con la presencia del fenómeno de patergia, refuerzan la hipótesis de un proceso inmunológico anormal, así como el de una respuesta inflamatoria descontrolada, exagerada y alterada2.
Se ha implicado tanto la inmunidad humoral como la celular. Las alteraciones humorales descritas incluyen (a) anticuerpos originados de una reacción cruzada entre antígenos del intestino y la piel (como la citoqueratina 18), cosa que justificaría las manifestaciones cutáneas en contexto de una EII, (b) un factor dermonecrótico del suero que induce necrosis cuando se inyecta en la piel del propio paciente, (c) un factor del suero de pacientes con PG que indujeron lesiones similares a PG cuando se inyectaron en cobayas8,9 y (d) se cree que podría existir una reacción cruzada entre antígenos bacterianos, principalmente de Escherichia coli y cutáneos, puesto que los tratamientos para disminuir la flora bacteriana intestinal han mostrado beneficios en el tratamiento del PG9–11.
Por otro lado, algunos estudios12 sugieren que el mecanismo patogénico podría ser una reacción de Arthus y Schwartzmann, dado que se han identificado inmunocomplejos en la pared de los vasos. Estudios de inmunoflurescencia y ultraestructurales han detectado depósito de IgG, IgM e IgA, así como componentes del complemento (C1q y C3). Se cree que estos inmunocomplejos depositados podrían activar las vías clásica y alternativa del complemento, dando lugar a la producción de anafilotoxinas C3a y C5a, las cuales inducen la degranulación de los mastocitos y atraen los neutrófilos. Estos se adhieren a las células endoteliales y migran al tejido conectivo para fagocitar y degradar los inmunocomplejos.
Además, se cree que existen defectos de hipersensibilidad o en la quimiotaxis de los neutrófilos, así como en la fagocitosis. Se ha descrito una variabilidad anormal en la migración y metabolismo oxigenado de los neutrófilos, así como un factor leucocitario que mejora la migración de los leucocitos sin alterar la actividad quimiotáctica2,13,14. Todo esto, sumado a que las vías para proteger la epidermis de la infiltración neutrofílica son insuficientes, resulta en una necrosis tisular1.
También se ha relacionado la interleucina 8 (IL-8) con la patogenia del PG. Oka et al.15 demostraron en su primer estudio una sobreexpresión de IL-8 en las úlceras de PG y que al inyectar un adenovirus recombinante que expresaba el ADN complementario codificante para IL-8 humana en ratones con inmunodeficiencia severa común, a los que se les había realizado un xenotrasplante de piel humana, estos desarrollaban unas úlceras que clínica e histológicamente eran muy parecidas a un PG, con lo que sugirieron que la IL-8 podría jugar un papel etiológico en la patogenia. Posteriormente, en otro estudio16 demostraron que la IL-8 era producida por los fibroblastos de la úlcera. Cultivaron fibroblastos de la úlcera y de piel sana, midiendo en diversas ocasiones los niveles de IL-8 en el sobrenadante durante 41 días. Paralelamente midieron los niveles plasmáticos de la misma interleucina durante 60 días. Vieron que los niveles en los fibroblastos aislados de la úlcera eran elevados durante los primeros días de cultivo y posteriormente, tras 27 días de cultivo, descendían rápidamente. Los niveles en los fibroblastos de piel sana fueron siempre indetectables y los plasmáticos fueron elevados en los primeros días y descendieron hasta hacerse indetectables a los 40 días de tratamiento. Con todo esto demostraron que la IL 8 es producida por los fibroblastos del PG y reforzaron la hipótesis en la que se implica la IL 8 en la patogenia del PG.
El papel de los defectos en la inmunidad celular no está del todo claro. Se han descrito múltiples alteraciones: anergia cutánea a Candida, estreptocinasa y derivados purificados de proteínas, así como una producción alterada de un factor inhibidor de macrófagos por parte de los linfocitos9. Se desconoce el papel exacto del linfocito T, pero se cree que este tiene que ser importante, puesto que la asociación a enfermedades sistémicas como la EII y la artritis reumatoide es frecuente, y en estas las células T son fundamentales. Algunos autores han identificado una expansión clonal de linfocitos T y neutrófilos en pacientes con PG. En el estudio realizado por Brooklyn et al.17 partieron de la hipótesis que las células T estimulan el proceso inflamatorio, con lo que sería lógico encontrar una expresión oligoclonal en la piel y en sangre periférica, dado que esas células responden a un antígeno. Demostraron que cinco pacientes con PG presentaban una clona de linfocitos T en sangre periférica y cuatro de ellos presentaban la misma clona en la piel.
Con esto concluyen que la célula T juega un papel integral en el PG y sugieren que los linfocitos T migran a la piel bajo la influencia de un estímulo antigénico.
Los factores que inician o mantienen estas alteraciones no están del todo claros, pero se cree que probablemente sea multifactorial, desde alteraciones genéticas a agentes infecciosos, e incluso alteraciones inmunológicas.
Recientemente, Nesterovitch et al.18 han descrito nuevas mutaciones, así como variantes aberrantes de acoplamiento genético en el gen PSTPIP 1, que codifica para la proteína homónima. Esta, llamada también CD2-binding proteína 1, se ha implicado en la organización del citoesqueleto. Hasta la actualidad se habían descrito dos mutaciones de sentido erróneo (A230T y E250Q) en este gen en pacientes afectos de síndrome PAPA (artritis estéril piogénica con PG y acné). Las nuevas mutaciones halladas (G258A y R52Q) se cree que pueden tener un efecto dañino sobre la función de la proteína. Las variantes aberrantes del acoplamiento generan una proteína truncada, lo que conlleva una pérdida de la función de la misma, aunque se desconoce la cantidad de proteína mutada necesaria para el desarrollo de síntomas clínicos. Por otro lado, demuestran que el número de repeticiones del tándem (CCTG)n en la región promotora de la proteína no aumenta el riesgo de desarrollar un PG.
Características clínicasEl PG es una entidad que puede presentarse en varias formas clínicas. Desde su primera descripción en 1916, se ha ido caracterizando tanto clínica como histológicamente, hasta llegar a la clasificación actual. Powell et al.9,19 describieron cuatro variantes principales.
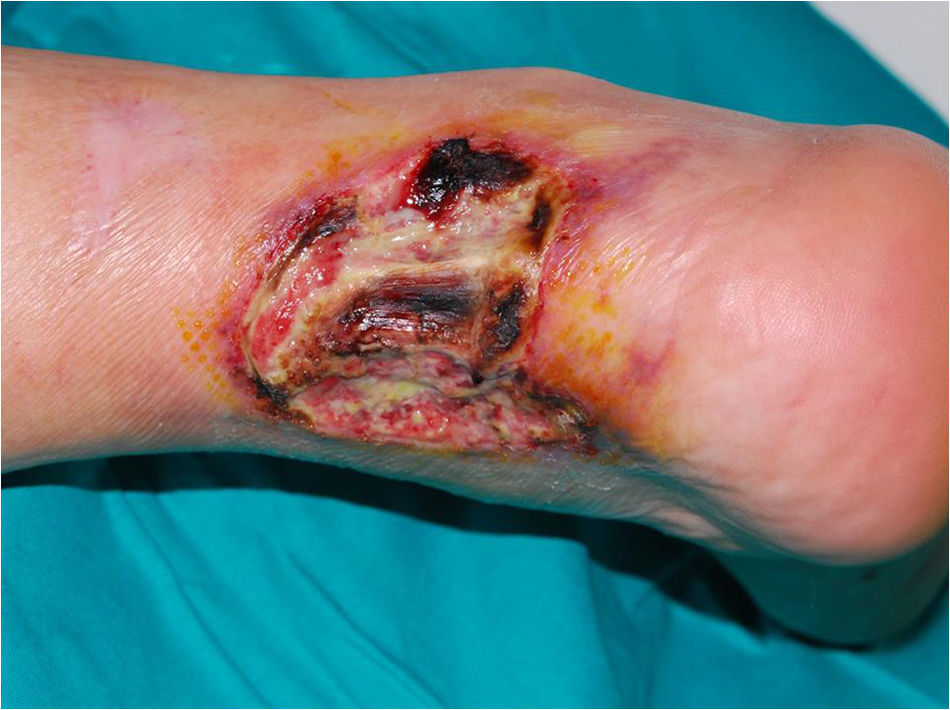
Variante clásica o ulcerativaEs la variante más frecuente y reconocida. Suele iniciarse bien como una pápula de pequeño tamaño, dolorosa y de coloración violácea, como un nódulo doloroso o como una pústula estéril, que rápidamente se extiende por los laterales con necrosis del tejido circundante y evoluciona a una úlcera dolorosa de bordes eritemato-violáceos, muy bien delimitados y socavados (figs. 1 y 2). En la base de la úlcera se suele encontrar tejido de granulación, necrosis o exudado purulento. La piel de alrededor habitualmente se encuentra eritematosa e indurada.


Típicamente el dolor es muy intenso, muchas veces desproporcionado a los hallazgos de la exploración física.
Las lesiones pueden ser únicas o múltiples, localizándose con más frecuencia en las extremidades inferiores, aunque pueden encontrarse en cualquier parte de la superficie corporal.
Algunos pacientes pueden presentar toxicidad y fiebre, así como mialgias y artralgias coincidiendo con el inicio de las lesiones. Con la instauración del tratamiento es frecuente observar una rápida respuesta; la epitelización empieza por los márgenes dejando una cicatriz atrófica y cribiforme, y en muchas ocasiones pigmentada.
Algunos autores distinguen dos subgrupos según el curso clínico: los que presentan una instauración explosiva con una rápida evolución de las lesiones, y los que la instauración es indolente y la evolución es más gradual2,14,20,21.
Variante pustulosaEs una variante infrecuente y superficial, descrita en 1978 por O’Loughlin y Perry22 como una forma abortiva de PG, que se presentaba como una erupción pustulosa, sin evolucionar a úlcera.
En esta variante se observa una erupción de pequeñas pústulas dolorosas con halo eritematoso alrededor, sobre piel sana, de predominio en tronco y superficies extensoras. Ocasionalmente estas lesiones pueden confluir y ulcerarse, dando lugar a lesiones persistentes y dolorosas. Típicamente se ha asociado a EII (sobre todo colitis ulcerosa) y especialmente en relación a brotes graves de la misma, junto con fiebre y artralgias. Suele observarse una mejoría de las lesiones con el tratamiento de la enfermedad subyacente2,9,23–25.
Existen algunos casos publicados de pacientes con EII en los que se han observado simultáneamente lesiones de PG pustuloso y ulcerativo. En ocasiones, el PG pustuloso se ha relacionado con la dermatosis pustulosa subcórnea26–28.
Dentro de esta variante se describió la Pyostomatitis vegetans. Se cree que es una forma de PG pustuloso oral, que se caracteriza por un edema y engrosamiento de la mucosa oral, junto con múltiples pústulas de 2-3mm de diámetro, así como úlceras en huella de caracol sobre una base eritematosa. Las lesiones orales frecuentemente se asocian a EII, especialmente a colitis ulcerosa activa9,29. El 50% de los casos presenta lesiones pustulosas cutáneas. Inicialmente, a los pocos años de su descripción, Hallopeau30 sugirió que esta entidad se trataba de una variante de pénfigo vegetante, dado que las lesiones se localizaban principalmente en flexuras y en el estudio histopatológico se observaba una importante eosinofilia tisular y en sangre periférica. Años después, numerosos autores publicaron casos de pacientes con esta variante, demostrando que el pioderma-piostomatitis vegetante es una entidad diferente y no relacionada con el pénfigo vegetante. Justificaron que tanto los hallazgos clínicos, como histopatológicos e inmunopatológicos los diferenciaban claramente31–33.
Dada esta controversia, estos autores consideran más apropiado hablar de una erupción asociada a EII más que de una forma de PG.
Variante ampollarEn 1972 Perry y Winkelmann describieron una nueva variante superficial de PG en tres pacientes con leucemia34, que se caracterizaba por ampollas y vesículas dolorosas e inflamatorias con un crecimiento concéntrico rápido, dando lugar a lesiones erosivas superficiales centrales. Dado que en esta entidad hay una necrosis superficial, gracias a la cual se da el crecimiento de las lesiones, es habitual observar una coloración grisácea en el tejido circundante.
Las localizaciones más frecuentes de esta variante son la cara y las extremidades superiores, y típicamente se ha asociado a enfermedades hematológicas, como por ejemplo leucemia mieloide aguda o a entidades mielodisplásicas, como la metaplasia mieloide.
Esta entidad comparte características clínicas e histopatológicas con el síndrome de Sweet y la dermatosis pustulosa subcórnea; por este motivo, hay autores que creen que en el contexto de enfermedades mieloproliferativas dichas entidades pueden representar diferentes presentaciones de un mismo espectro de condiciones reactivas cutáneas9,19,35.
El pronóstico está marcado por la enfermedad subyacente, por lo que suele ser bastante pobre2,24.
Variante vegetativaEs una variante superficial, localizada, limitada y poco agresiva de PG, que fue descrita por Wilson-Jones et al.36 en el año 1988, bajo el nombre de pioderma granulomatoso superficial. Estos autores publicaron una serie de 25 pacientes que presentaban una variante de pioderma superficial, ulcerativa y vegetante, con formación de granulomas en el estudio histológico y que en su gran mayoría curaban sin necesidad de tratamiento sistémico.
Clínicamente se presenta como una úlcera superficial, localizada principalmente en el tronco, con una base no purulenta y con unos bordes que no muestran las características típicas de la forma ulcerativa. En este caso, los bordes no suelen ser violáceos ni socavados y, además, no suele observarse inflamación circundante. En la mayoría de casos no se asocia a ninguna enfermedad subyacente. Un dato importante que nos orienta sobre su escasa agresividad y nos ayuda al diagnóstico, es la respuesta favorable a los tratamientos tópicos2,5,9,24.
Otras variantesExisten varias formas de PG, que se clasifican según la localización anatómica, ya sea porque presentan características específicas que pueden ayudar al diagnóstico, porque presentan algunas enfermedades típicamente asociadas o porque tienen un tratamiento específico.
Variante periostomalEsta forma representa casi el 15% del total de los PG24, con una incidencia anual de un 0,6% entre los pacientes portadores de ostomías –siendo la más frecuente la ileostomía (3 de 500 pacientes con ostomías en un estudio realizado por Lyon et al.37)-. Es una entidad que típicamente se ha asociado a EII, aunque recientemente también se ha observado asociada a enfermedad diverticular intestinal, neoplasias intestinales, perforación intestinal, vejiga neurógena, colitis colágena y esclerosis sistémica2,4,38–40.
Se caracteriza por la presencia de úlceras similares a las del PG clásico alrededor de la ostomía, en las que es frecuente encontrar puentes de piel sana que atraviesan la base de la úlcera. Las lesiones son dolorosas y habitualmente interfieren con la normal adherencia de las bolsas colectoras23,24. Se ha observado una latencia de entre 2 meses a 25 años después de la intervención. Se cree que en esta variante el fenómeno de patergia juega un papel importante, representando en este caso el factor traumático la irritación continua por las heces, los adhesivos de las bolsas o los desbridamientos2.
Variante genitalLa característica principal, que diferencia esta variante de la clásica, es la localización, puesto que clínicamente no difiere. Es una forma bastante infrecuente. Se ha descrito la forma vulvar y la peneana, siendo la localización en el escroto más rara. Esta variante es típica de la edad infantil. En un estudio realizado por Graham et al.41, en el que revisaron los PG infantiles publicados en la literatura inglesa, vieron que en la población infantil había una distribución inusual en la zona perianal y genital, las cuales no son típicas en la edad adulta.
El diagnóstico definitivo es difícil, motivo por el cual suele ser necesaria la realización de múltiples biopsias, así como la presencia de múltiples cultivos negativos19.
Cuando existen lesiones genitales, se tiene que hacer el diagnóstico diferencial con la enfermedad de Behçet. En esta última entidad es frecuente encontrar, a parte de las lesiones aftoides genitales, otras características típicas de la enfermedad2,23,42,43.
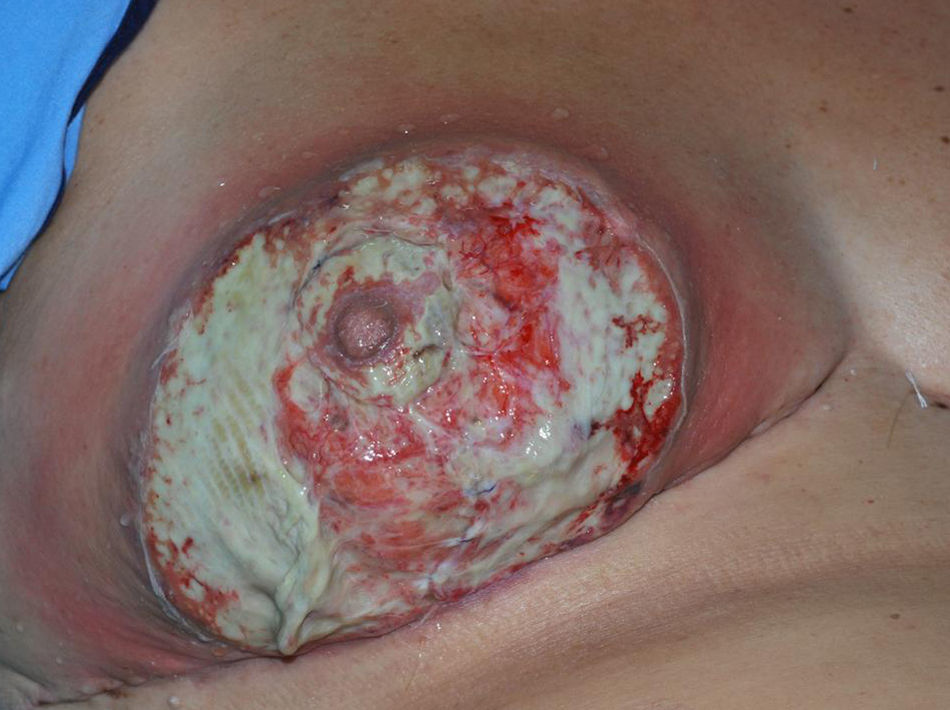
Variante localizada en las mamasLa afectación de la mama es infrecuente. Existen unos 400 casos descritos en la literatura. Las características clínicas de las lesiones son muy similares a las del PG clásico, aunque típicamente la úlcera respeta el pezón, hecho que es de mucha utilidad en el diagnóstico (fig. 3). En la mayoría de casos publicados las lesiones iban precedidas de alguna intervención quirúrgica, probablemente debido a un fenómeno de patergia44–48. Este hecho es muy importante, dado que a las pacientes que ya hayan desarrollado alguna lesión de PG previamente se les tiene que desaconsejar las intervenciones que no sean estrictamente necesarias. La intervención quirúrgica que más se ha relacionado con PG mamario es la mastoplastia de reducción, probablemente porque las mamas hipertróficas presentan un aporte sanguíneo más precario y, además, el grado de traumatismo es mayor que en otras intervenciones. Otro dato relevante es que esta es una de las intervenciones mamarias más frecuentes49,50.

El principal diagnóstico diferencial se tiene que establecer con infecciones cutáneas y del tejido subcutáneo, tales como la fascitis necrotizante y neoplasias malignas4,23.
Variante infantilEl PG infantil representa entre el 4-5% del total, con una predilección por la zona perineal, cabeza, cara, cuello y piernas. En la mayoría de casos la respuesta al tratamiento suele ser satisfactoria21,41.
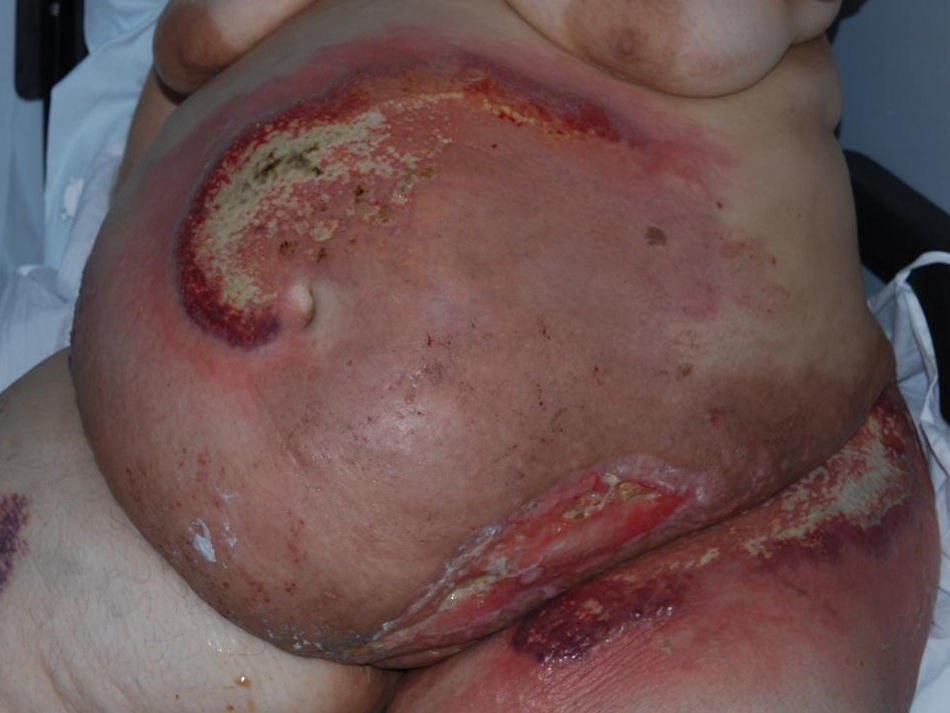
Variante posquirúrgicaEl PG posquirúrgico se puede considerar una variante aparte, dado que posee características específicas. Inicialmente suele aparecer una zona eritematosa alrededor de la herida quirúrgica, seguida de una ulceración y necrosis cutánea (figs. 2 y 3). El tiempo de latencia entre la intervención y la aparición de la lesión puede ser muy variable y depende principalmente de la intensidad del traumatismo. De este modo, se ha observado que la latencia después de una biopsia es de entre 3 y 4 meses, en cambio el PG que aparece tras una cirugía mayor lo hace con una latencia de entre 4 y 40 días49. En la mayoría de los casos, inicialmente se diagnostica de infección posquirúrgica y se trata con antibióticos de amplio espectro e incluso oxígeno hiperbárico. Ante la ausencia de mejoría, en ocasiones se realiza desbridamiento quirúrgico que puede empeorar aún más la lesión por el fenómeno de patergia. El diagnóstico suele ser de exclusión de las otras posibles causas, observándose retrasos en el diagnóstico que van de los 10 días a los 28 meses50.
Scott et al.50 sugieren que el hipotiroidismo podría ser una comorbilidad que predispusiera a la aparición de PG posquirúrgico, dado que observaron esta enfermedad en el 28% de sus pacientes con PG posquirúrgico.
Es importante la realización de cultivos microbiológicos, puesto que pueden ayudar al diagnóstico. En los casos en los que, dada la sospecha diagnóstica inicial de infección posquirúrgica, se hayan tratado con antibióticos y los cultivos sean repetidamente negativos, un buen método para excluir el posible origen infeccioso es la secuenciación del ARN(r) ribosómico de la subunidad 16S51.
Así pues, es importante que ante una úlcera posquirúrgica de rápida instauración, dolorosa y con cultivos microbiológicos negativos, se sospeche desde un inicio esta entidad, se tome una biopsia y se instaure el tratamiento correcto para evitar secuelas importantes.
Fenómeno de patergiaEl 20-40% de los casos presenta fenómeno de patergia5,9,23,24,52–54. Este fenómeno se refiere a la aparición de una lesión de PG posterior a un traumatismo, en el sitio de aplicación. Se ha descrito, entre otros, tras una intervención quirúrgica (el caso del PG en las mamas es un claro ejemplo), una biopsia o tras un traumatismo banal. Este hecho nos es de gran utilidad, en muchas ocasiones, para orientar el diagnóstico de las úlceras. En este sentido, se tiene que recomendar al paciente que evite traumatismos o intervenciones quirúrgicas innecesarias. En los casos en los que la cirugía no se pueda evitar, existen algunas recomendaciones que se deberían seguir: (a) en el momento de la intervención el paciente tendría que estar en tratamiento corticoideo, (b) este se debería mantener un mínimo de 6 meses después de la intervención y, por último, (c) se tendrían que evitar las suturas47,48.
Manifestaciones extracutáneasAunque la afectación visceral por PG es infrecuente, se han descrito numerosas manifestaciones extracutáneas. La más frecuente son los infiltrados neutrofílicos estériles viscerales, aunque también se ha descrito la formación de abscesos. El órgano más frecuentemente afectado es el pulmón. Asimismo, se han publicado casos de osteomielitis, adenitis, abscesos esplénicos y hepatopancreáticos4,19,55–58.
Esta afectación extracutánea puede preceder, aparecer al mismo momento que el PG o hacerlo posteriormente. Por este motivo el diagnóstico clínico de esta afectación no es fácil. Así, las características clínicas, los hallazgos histopatológicos en la biopsia cutánea, los cultivos repetidamente negativos, la falta de respuesta a antibióticos y la rápida mejoría con los corticosteroides sistémicos pueden ser de gran ayuda.
HistopatologíaDebido a que los hallazgos histopatológicos del PG no son específicos y suelen ser bastante variables, el diagnóstico definitivo de la entidad recae principalmente en las características clínicas. El objetivo principal de la biopsia es descartar las otras posibles causas de úlceras.
Es muy importante tener en cuenta que la histología del PG puede variar según el tipo de lesión, el sitio de donde se obtiene la muestra, el estadio evolutivo de la lesión y si ya se ha iniciado tratamiento o no.
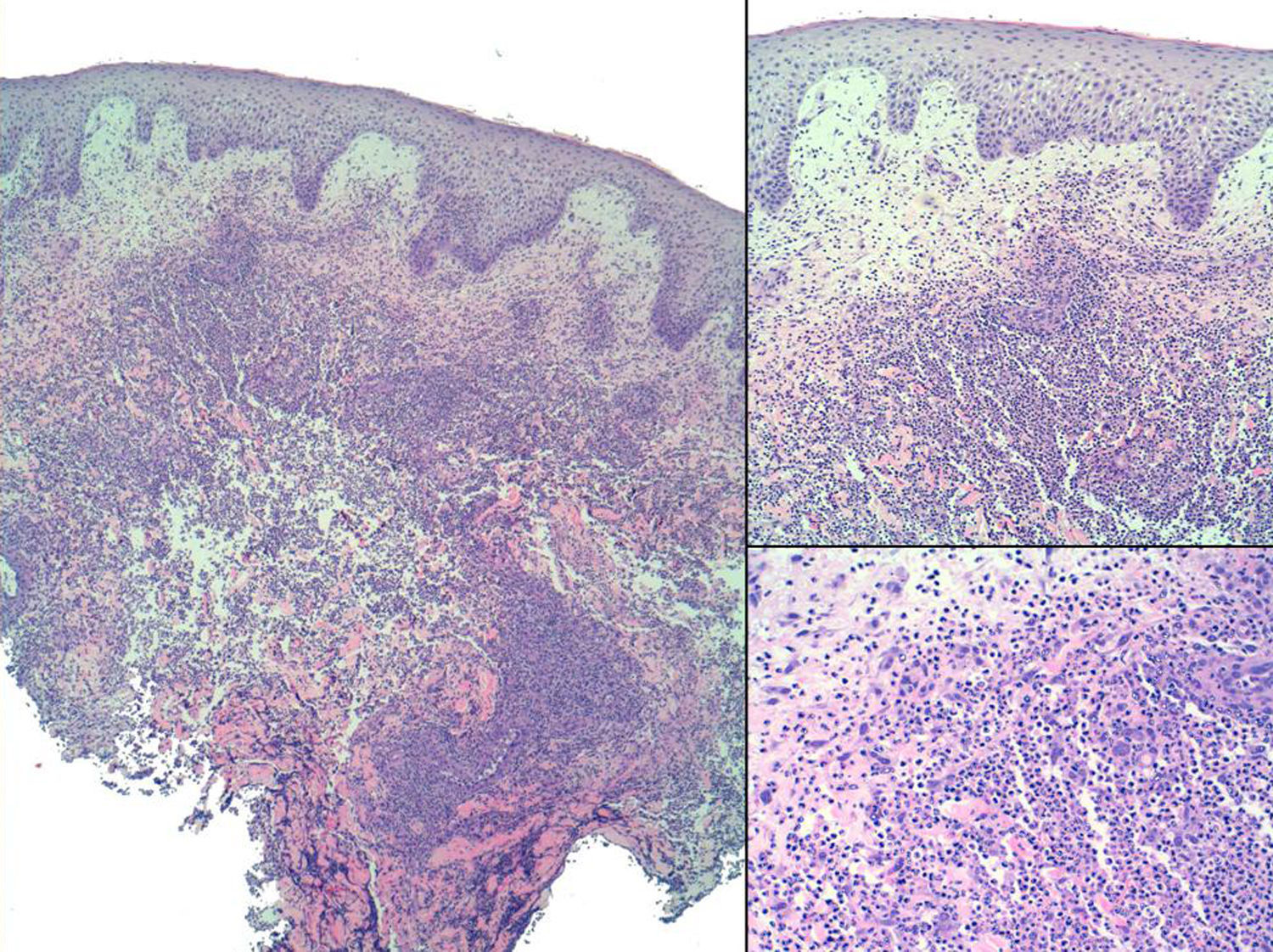
Lo más recomendable es hacer una biopsia de los márgenes necróticos y socavados, dado que suelen encontrarse hallazgos que nos pueden ser de gran ayuda. Típicamente se observa una necrosis central y ulceración de la epidermis y dermis, con un infiltrado inflamatorio intenso y agudo, así como un infiltrado periférico que presenta características mixtas con predominancia de neutrófilos (fig. 4). Powell et al.9 describieron las diferencias en los infiltrados inflamatorios según el sitio del cual se tomaba la biopsia. En el centro, base o borde de la úlcera suele encontrarse un infiltrado predominantemente neutrofílico, que tiende a la formación de abscesos. Más a la periferia se suele encontrar un infiltrado mixto y finalmente, en la areola eritematosa que rodea la úlcera es habitual encontrar un infiltrado linfocítico que se concentra alrededor de los vasos, pudiéndose observar daño vascular. Algunos autores han descrito la presencia de vasculitis en las muestras estudiadas, demostrando en la inmunofluorescencia directa un depósito vascular de IgM, C3 y fibrina23,59,60, aunque estos hallazgos no han podido ser demostrados por otros autores. Se cree que en los casos en los que la vasculitis es prominente, se tendría que considerar como primer diagnóstico una vasculopatía primaria23,52.

En las lesiones iniciales es frecuente observar infiltrados neutrofílicos, mientras que en las úlceras ya evolucionadas se observa una marcada necrosis tisular con un infiltrado mononuclear alrededor. Las lesiones en regresión o ya curadas muestran una importante fibroplasia61.
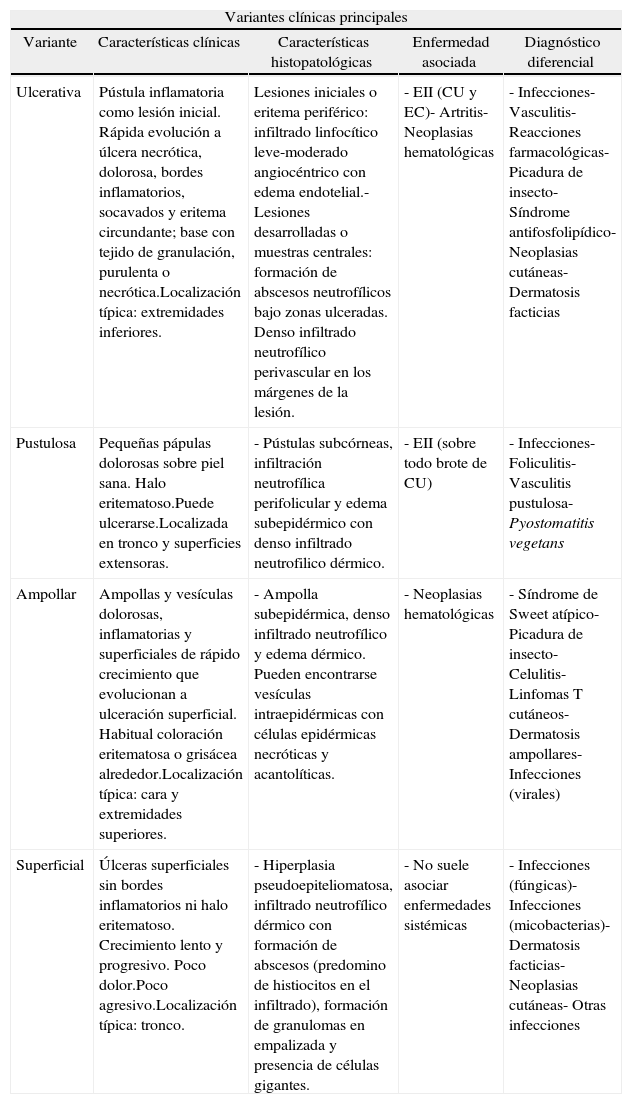
Cada variante clínica tiene hallazgos histopatológicos más específicos que ayudan a su diagnóstico (tabla 1).
Variantes clínicas del PG y sus principales características
| Variantes clínicas principales | ||||
| Variante | Características clínicas | Características histopatológicas | Enfermedad asociada | Diagnóstico diferencial |
| Ulcerativa | Pústula inflamatoria como lesión inicial. Rápida evolución a úlcera necrótica, dolorosa, bordes inflamatorios, socavados y eritema circundante; base con tejido de granulación, purulenta o necrótica.Localización típica: extremidades inferiores. | Lesiones iniciales o eritema periférico: infiltrado linfocítico leve-moderado angiocéntrico con edema endotelial.- Lesiones desarrolladas o muestras centrales: formación de abscesos neutrofílicos bajo zonas ulceradas. Denso infiltrado neutrofílico perivascular en los márgenes de la lesión. | - EII (CU y EC)- Artritis- Neoplasias hematológicas | - Infecciones- Vasculitis- Reacciones farmacológicas- Picadura de insecto- Síndrome antifosfolipídico- Neoplasias cutáneas- Dermatosis facticias |
| Pustulosa | Pequeñas pápulas dolorosas sobre piel sana. Halo eritematoso.Puede ulcerarse.Localizada en tronco y superficies extensoras. | - Pústulas subcórneas, infiltración neutrofílica perifolicular y edema subepidérmico con denso infiltrado neutrofilico dérmico. | - EII (sobre todo brote de CU) | - Infecciones- Foliculitis- Vasculitis pustulosa- Pyostomatitis vegetans |
| Ampollar | Ampollas y vesículas dolorosas, inflamatorias y superficiales de rápido crecimiento que evolucionan a ulceración superficial. Habitual coloración eritematosa o grisácea alrededor.Localización típica: cara y extremidades superiores. | - Ampolla subepidérmica, denso infiltrado neutrofílico y edema dérmico. Pueden encontrarse vesículas intraepidérmicas con células epidérmicas necróticas y acantolíticas. | - Neoplasias hematológicas | - Síndrome de Sweet atípico- Picadura de insecto- Celulitis- Linfomas T cutáneos- Dermatosis ampollares- Infecciones (virales) |
| Superficial | Úlceras superficiales sin bordes inflamatorios ni halo eritematoso. Crecimiento lento y progresivo. Poco dolor.Poco agresivo.Localización típica: tronco. | - Hiperplasia pseudoepiteliomatosa, infiltrado neutrofílico dérmico con formación de abscesos (predomino de histiocitos en el infiltrado), formación de granulomas en empalizada y presencia de células gigantes. | - No suele asociar enfermedades sistémicas | - Infecciones (fúngicas)- Infecciones (micobacterias)- Dermatosis facticias- Neoplasias cutáneas- Otras infecciones |
| Otras variantes | |
| Variante | Comentario |
| Periostomal | Úlceras similares al clásico, alrededor de ostomías con tractos de piel sana que atraviesan la base. Latencia variable. Patergia por irritación continua y adhesivos de las bolsas. |
| Genital | Típica de niños. Clínica similar a la forma clásica. Forma vulvar y peneana. Escrotal infrecuente. Diagnóstico diferencial con enfermedad de Behçet. |
| Localizado en mamas | Infrecuente. Típicamente post-traumática (intervención quirúrgica, sobre todo mastoplastia de reducción). Diagnóstico diferencial con infecciones post-quirúrgicas de partes blandas y neoplasias. |
| Infantil | 4-5% del total. Zona perineal, cabeza, cara, cuello y piernas. Mayoría de casos buen pronóstico. |
Entre el 50 y 60% de los pacientes con PG presenta alguna patología asociada. Las más frecuentes son la EII, la artritis, la paraproteinemia y las neoplasias hematológicas. Muchas otras también se han relacionado, pero de forma mucho menos frecuente21 (tabla 2).
Enfermedades asociadas a PG
| Enfermedades asociadas | |
| Enfermedades habitualmente asociadas | Enfermedad inflamatoria intestinal (colitis ulcerosa y enfermedad de Crohn)Artritis (seropositiva o seronegativa, monoarticular o poliarticular, simétrica o asimétrica)ParaproteinemiasNeoplasias hematológicas (leucemia aguda o crónica de estirpe mieloide, mielofibrosis, metaplasia mieloide, leucemia de células peludas) |
| Otras enfermedades asociadas | Tracto gastrointestinal (diverticulosis, hepatitis crónica activa, cirrosis biliar primaria)Artritis (espondilitis anquilosante, osteoartritis, policondritis, artritis psoriásica)Alteraciones hematológicas (policitemia vera, mieloma múltiple, hipergammaglobulinemia congénita y adquirida, macroglobulinemia de Waldenström, linfomas, esplenomegalia y hemoglobinuria paroxística nocturna)Enfermedades del tejido connectivo (enfermedad de Takayasu, lupus eritematoso sistémico, granulomatosis de Wegener y vasculitis necrotizante)Neoplasias (múltiples neoplasias sólidas –colon, próstata, mama y pulmón y tumor carcinoide–)Otras (hidrosadenitis supurativa, acne conglobata, VIH, diabetes, enfermedades tiroideas, enfermedad pulmonar obstructiva crónica) |
Los pacientes con EII representan el 15-20% de todos los PG y se estima que el 0,75-5% de los pacientes con EII presentan en algún momento de la enfermedad PG.
Inicialmente se creía que el PG era más frecuente entre pacientes con colitis ulcerosa (CU), pero en estudios posteriores se vio que la incidencia era muy parecida en la CU y la enfermedad de Crohn (EC). La actividad de la EII y del PG siguen cursos independientes, pudiendo aparecer manifestaciones cutáneas en cualquier punto evolutivo de la enfermedad intestinal, incluso después de la resección quirúrgica del intestino inflamado. No obstante, en algunos pacientes los síntomas de la EII preceden a la aparición de PG e incluso las exacerbaciones intestinales se relacionan con un empeoramiento de la patología cutánea2,4,6,9,23. La patología intestinal se relaciona sobre todo con las variantes clásica y pustulosa. El mecanismo por el cual se asocian es desconocido, pero se cree que podría existir una reacción cruzada entre Eschericha coli y algunos antígenos cutáneos. Algunos autores creen que esta bacteria podría tener alguna implicación en el mecanismo inflamatorio implicado la CU y en el PG9,11.
ArtritisLa artritis se asocia frecuentemente. Powell et al. describieron en su serie de 86 pacientes una prevalencia de un 37% en los que presentaban un PG ulcerativo. La forma de presentación más frecuentemente descrita es la de una monoartritis seronegativa asimétrica2,9, aunque hay autores que describen la poliartritis simétrica seronegativa como la más frecuente23. Otros autores, como Bennett y Saracino21,62, presentan series de casos en los que lo más frecuente es la artritis reumatoide8. Por último, hay algunos que sugieren que esta afectación podría ser una manifestación extraintestinal de una EII, dada la alta frecuencia con la que se relacionan la EII y la artritis en paciente con PG23. Otras formas descritas menos frecuentes incluyen la artritis psoriásica, síndrome de Felty, osteoartritis, sacroileítis y espondilitis, esta última en el contexto de EII. Hay algún caso descrito de PG asociado a síndrome de SAPHO63,64.
Normalmente la afectación articular precede a la cutánea, aunque el curso de las dos enfermedades son independientes.
ParaproteinemiasHasta el 15% de los PG presentan una paraproteinemia asociada, normalmente benigna. Lo más frecuente es que se trate de una IgA, aunque casos de IgG e IgM también se han descrito.
Se ha observado que la IgA inhibe la función neutrofílica in vitro, por este motivo se cree que podría favorecer un entorno inmunológico para el desarrollo de PG.
La evolución a mieloma múltiple (MM) es infrecuente. En los casos en los que se da, la aparición de MM suele ser posterior a la del PG. Por este motivo es recomendable hacer un seguimiento estrecho de estos pacientes.
La variante que se asocia con más frecuencia es la ulcerativa, aunque la ampollar también es frecuente, sobre todo en pacientes con MM2,9,21,23,65,66.
Neoplasias hematológicasRepresentan entre el 7-25% de los casos de PG. Las leucemias de estirpe mieloide son las más frecuentemente asociadas, ya sea aguda (LAM), crónica (LMC) o mielomonocítica (LMM). También se han descrito con síndromes mielodisplásicos, como la metaplasia mieloide, la anemia refractaria con exceso de blastos y la tricoleucemia. Otras neoplasias descritas, pero de modo menos frecuente, incluyen mielofibrosis, linfoma de Hodgkin, linfoma no Hodgkin, linfomas cutáneos T, macroglobulinemia de Waldenström, MM y policitemia rubra vera. Esta última entidad se ha descrito sobre todo en niños y la aparición de PG en su contexto se asocia a mal pronóstico, dado que en un alto porcentaje de los pacientes sufren una transformación a leucemia en los meses siguientes al diagnóstico67. Estas enfermedades se asocian con más frecuencia en las variantes atípicas, sobre todo la ampollar o bien en pacientes que a la vez presentan características típicas de síndrome de Sweet2,4,9,21,23.
Otros procesos asociadosSe han identificado múltiples enfermedades, no incluidas en las secciones anteriores, asociadas a PG. Las alteraciones inmunológicas incluyen defectos en la inmunidad celular y humoral, como la hipogammaglobulinemia congénita o adquirida y el síndrome de hiperinmunoglobulina E. La hepatitis crónica activa, la cirrosis biliar primaria, el lupus eritematoso sistémico, las enfermedades tiroideas, el acné conglobata y la sarcoidosis, entre muchas otras enfermedades, también se han observado en asociación a PG2,9,20,23,68. (tabla 2)
Recientemente, en un revisión realizada por Binus et al. en 103 pacientes, identificaron una alta incidencia de hepatitis viral o autoinmune (8,5%) y depresión (21,5%), concluyendo que estas dos entidades se tendrían que incluir en las enfermedades típicamente asociadas o que al menos se tendrían que tener en cuenta a la hora de hacer el estudio de estos pacientes69.
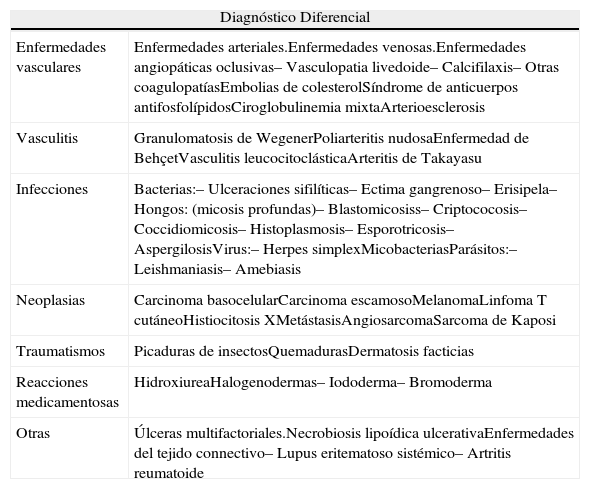
Diagnóstico diferencialEl diagnóstico diferencial del PG incluye todas las enfermedades que puedan causar úlceras cutáneas. Entre estas se encuentran: úlceras de origen vascular, que son las más frecuentes, vasculitis, enfermedades venosas isquémicas, ulceras facticias, neoplasias, traumatismos, infecciones, enfermedades metabólicas, hematológicas y causas relacionadas con fármacos. (tabla 3)
Diagnóstico diferencial del PG
| Diagnóstico Diferencial | |
| Enfermedades vasculares | Enfermedades arteriales.Enfermedades venosas.Enfermedades angiopáticas oclusivas– Vasculopatia livedoide– Calcifilaxis– Otras coagulopatíasEmbolias de colesterolSíndrome de anticuerpos antifosfolípidosCiroglobulinemia mixtaArterioesclerosis |
| Vasculitis | Granulomatosis de WegenerPoliarteritis nudosaEnfermedad de BehçetVasculitis leucocitoclásticaArteritis de Takayasu |
| Infecciones | Bacterias:– Ulceraciones sifilíticas– Ectima gangrenoso– Erisipela– Hongos: (micosis profundas)– Blastomicosiss– Criptococosis– Coccidiomicosis– Histoplasmosis– Esporotricosis– AspergilosisVirus:– Herpes simplexMicobacteriasParásitos:– Leishmaniasis– Amebiasis |
| Neoplasias | Carcinoma basocelularCarcinoma escamosoMelanomaLinfoma T cutáneoHistiocitosis XMetástasisAngiosarcomaSarcoma de Kaposi |
| Traumatismos | Picaduras de insectosQuemadurasDermatosis facticias |
| Reacciones medicamentosas | HidroxiureaHalogenodermas– Iododerma– Bromoderma |
| Otras | Úlceras multifactoriales.Necrobiosis lipoídica ulcerativaEnfermedades del tejido connectivo– Lupus eritematoso sistémico– Artritis reumatoide |
El diagnóstico de PG, como ya se ha comentado anteriormente, se basa en las características clínicas y la exclusión de otras posibles etiologías, dado que la histopatología no muestra hallazgos característicos y no existen marcadores serológicos o hematológicos definitorios.
Es muy importante realizar una historia clínica detallada, atendiendo a los antecedentes patológicos personales y familiares, a las características y evolución de la lesión, a la presencia de algún traumatismo anterior, los posibles síntomas asociados e historia de ingesta de fármacos. Por otro lado, es muy recomendable tomar una biopsia para descartar otras etiologías. Idealmente se tendría que hacer una biopsia en huso del borde inflamatorio para tinción de hematoxilina-eosina, Gram y plata metenamina y otro huso del margen de la úlcera para cultivo para descartar infecciones bacterianas, fúngicas, parasitarias y por micobacterias atípicas. Es importante realizar estudios gastrointestinales en pacientes seleccionados a través de la historia clínica. Esos incluyen sigmoidoscopia y/o colonoscopia, enemas baritados o radiografías seriadas.
La realización de estudios hematológicos, incluyendo hemograma completo, velocidad de sedimentación globular (VSG), bioquímica, proteinograma, crioglobulinas, anticuerpos anticitoplasma de neutrófilos (ANCA), así como estudios de coagulación (tiempo de protrombina, tiempo de tromboplastina parcial y anticuerpos anticardiolipina, entre otros), nos puede ayudar a descartar neoplasias hematológicas, paraproteinemias, vasculitis y síndrome de anticuerpos antifosfolípidos.
Es necesario el seguimiento estrecho de estos pacientes. Se tiene que monitorizar la respuesta y la aparición de posibles efectos secundarios al tratamiento, y replantearse el diagnóstico ante la ausencia de respuesta al tratamiento. Además, es necesario un seguimiento a largo plazo para descartar la posible presencia de alguna de las enfermedades asociadas2,5,9,70.
TratamientoEl objetivo principal del tratamiento es la eliminación completa de la actividad inflamatoria, la curación de la herida, el control del dolor y de las posibles enfermedades asociadas. La rápida instauración del tratamiento evitará la progresión de la lesión y disminuirá el riesgo de la aparición de grandes cicatrices antiestéticas. No existen estudios controlados que evalúen la eficacia del tratamiento. Por este motivo la mayoría de pautas de tratamiento se basan en series de casos, experiencias personales y opiniones de expertos. El tipo de tratamiento seleccionado dependerá de la extensión de la lesión, la profundidad, la evolución, el número de lesiones, las posibles patologías asociadas y el estado basal del paciente.
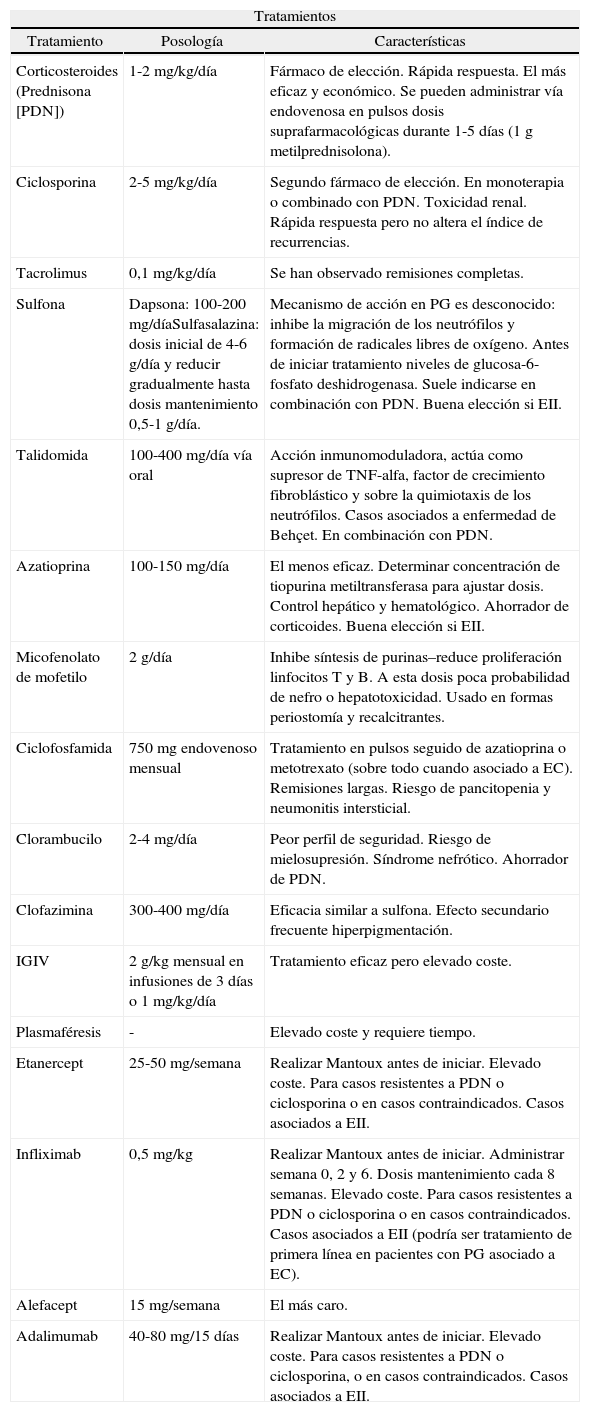
El tratamiento tópico puede ser una buena opción para los casos leves o moderados y no asociados a otras enfermedades. No obstante, también son muy importantes como terapia adyuvante en el tratamiento de los casos más severos o recalcitrantes. El tratamiento sistémico es de elección en casos severos o con patologías de base asociada. (tabla 4)
Principales tratamientos sistémicos del PG, su posología y principales características
| Tratamientos | ||
| Tratamiento | Posología | Características |
| Corticosteroides (Prednisona [PDN]) | 1-2mg/kg/día | Fármaco de elección. Rápida respuesta. El más eficaz y económico. Se pueden administrar vía endovenosa en pulsos dosis suprafarmacológicas durante 1-5 días (1g metilprednisolona). |
| Ciclosporina | 2-5mg/kg/día | Segundo fármaco de elección. En monoterapia o combinado con PDN. Toxicidad renal. Rápida respuesta pero no altera el índice de recurrencias. |
| Tacrolimus | 0,1mg/kg/día | Se han observado remisiones completas. |
| Sulfona | Dapsona: 100-200mg/díaSulfasalazina: dosis inicial de 4-6g/día y reducir gradualmente hasta dosis mantenimiento 0,5-1g/día. | Mecanismo de acción en PG es desconocido: inhibe la migración de los neutrófilos y formación de radicales libres de oxígeno. Antes de iniciar tratamiento niveles de glucosa-6-fosfato deshidrogenasa. Suele indicarse en combinación con PDN. Buena elección si EII. |
| Talidomida | 100-400mg/día vía oral | Acción inmunomoduladora, actúa como supresor de TNF-alfa, factor de crecimiento fibroblástico y sobre la quimiotaxis de los neutrófilos. Casos asociados a enfermedad de Behçet. En combinación con PDN. |
| Azatioprina | 100-150mg/día | El menos eficaz. Determinar concentración de tiopurina metiltransferasa para ajustar dosis. Control hepático y hematológico. Ahorrador de corticoides. Buena elección si EII. |
| Micofenolato de mofetilo | 2g/día | Inhibe síntesis de purinas–reduce proliferación linfocitos T y B. A esta dosis poca probabilidad de nefro o hepatotoxicidad. Usado en formas periostomía y recalcitrantes. |
| Ciclofosfamida | 750mg endovenoso mensual | Tratamiento en pulsos seguido de azatioprina o metotrexato (sobre todo cuando asociado a EC). Remisiones largas. Riesgo de pancitopenia y neumonitis intersticial. |
| Clorambucilo | 2-4mg/día | Peor perfil de seguridad. Riesgo de mielosupresión. Síndrome nefrótico. Ahorrador de PDN. |
| Clofazimina | 300-400mg/día | Eficacia similar a sulfona. Efecto secundario frecuente hiperpigmentación. |
| IGIV | 2g/kg mensual en infusiones de 3 días o 1mg/kg/día | Tratamiento eficaz pero elevado coste. |
| Plasmaféresis | - | Elevado coste y requiere tiempo. |
| Etanercept | 25-50mg/semana | Realizar Mantoux antes de iniciar. Elevado coste. Para casos resistentes a PDN o ciclosporina o en casos contraindicados. Casos asociados a EII. |
| Infliximab | 0,5mg/kg | Realizar Mantoux antes de iniciar. Administrar semana 0, 2 y 6. Dosis mantenimiento cada 8 semanas. Elevado coste. Para casos resistentes a PDN o ciclosporina o en casos contraindicados. Casos asociados a EII (podría ser tratamiento de primera línea en pacientes con PG asociado a EC). |
| Alefacept | 15mg/semana | El más caro. |
| Adalimumab | 40-80mg/15 días | Realizar Mantoux antes de iniciar. Elevado coste. Para casos resistentes a PDN o ciclosporina, o en casos contraindicados. Casos asociados a EII. |
Las curas locales de las úlceras juegan un papel importante. La utilización de apósitos que retienen la humedad ha demostrado ser superior a las curas con gasas secantes, puesto que dan un mejor control del dolor, inducen la producción de colágeno, facilitan el desbridamiento autolítico y promueven la angiogénesis71. Además, estos apósitos son más efectivos en la prevención de sobreinfecciones que las gasas. Los apósitos bioclusivos semipermeables también han demostrado ser efectivos23. Hay casos en los que el exudado es muy abundante y en vez de utilizar uno que favorezca la maceración, como serían lo oclusivos, se tendría que optar por los alginatos o hidrofibras, que son sustancias muy absorbentes72.
Es importante realizar cultivos de la lesión si se observa celulitis o linfangitis, puesto que la sobreinfección es una complicación posible y frecuente. En estos casos se tendría que instaurar la antibioticoterapia adecuada según antibiograma.
Se desaconseja el desbridamiento de las lesiones, puesto que por el fenómeno de patergia las úlceras pueden empeorar. Los injertos, en general, también están contraindicados, dado que es frecuente la aparición de lesiones de PG en la zona dadora.
Los principios activos que han demostrado efectividad como agentes tópicos son los corticoides potentes (tópicos o intralesionales), el tacrolimus y la ciclosporina (intralesional)2,4,23,71,73. Marzano et al.74 presentaron una serie de 8 casos de pacientes con PG localizados de los cuales 5 habían remitido completamente a las 6 semanas y sugirieron que el tacrolimus tópico en monoterapia podría ser un tratamiento de primera línea en pacientes con PG localizado, idiopático y de reciente instauración con cultivos microbiológicos negativos.
Existen casos tratados favorablemente con oxígeno hiperbárico, peróxido de benzoílo, nicotina y cromoglicato disódico.
Tratamientos sistémicosLos corticoides sistémicos a dosis de 1-2mg/kg/día se consideran el tratamiento de elección en el PG idiopático. Dado que la duración del tratamiento en estos pacientes suele ser de varios meses e incluso años, la probabilidad de aparición de efectos secundarios es elevada (se dan en un 50% de los pacientes tratados2). En estos casos se pueden asociar con otros inmunosupresores, que actúan como fármacos ahorradores de corticoides. También se indican en casos refractarios.
La ciclosporina A es el segundo fármaco más utilizado en el PG, ya sea en monoterapia o asociado a corticosteroides. Es un fármaco que tiene entre sus efectos secundarios la toxicidad renal, por lo que se tendría que indicar solo en los casos con PG idiopático, dado que no es apropiada para tratamientos de larga duración, como requeriría un paciente con EII2,4,71. Generalmente cuando se disminuye la dosis de este fármaco, es necesario añadir otros agentes sistémicos para mantener la respuesta. La dapsona, sulfapiridina y sulfasalazina son fármacos útiles, siendo este último el más eficaz2.
Otros fármacos como el metotrexato, la clofazimina, la talidomida, azatioprina, clorambucilo, ciclofosfamida y el micofenolato de mofetilo, entre otros, han sido utilizados con éxito en algunos pacientes4,9,21,23,71,75–78. Algunos autores han utilizado con éxito la plasmaféresis y la inmunoglobulina intravenosa (IGIV)79–82.
Recientemente, desde el año 2007, se han empezado a utilizar los inhibidores del factor de necrosis tumoral alfa (TNF-α), principalmente en los casos de PG asociados a EII. Se han publicado múltiples series de casos tratados con infliximab, adalimumab, etanercept, efalizumab y con alefacept. En algunos de los casos publicados, los pacientes no presentaban EII, pero se trataba de PG resistentes a otros fármacos sistémicos4,83–88.
Brooklyn et al.89 realizaron un estudio aleatorizado, a doble ciego y controlado realizado en 30 pacientes, en el que se evaluaba la eficacia de infliximab en PG. Observaron una tasa de respuesta del 69%, con un 21% de pacientes con remisión completa a las 6 semanas de tratamiento.
Loricnz et al.90 presentaron el caso de un paciente con EII y PG al que se le administró visilizumab (anticuerpo monoclonal que se une a la cadena épsilon del CD3 expresado en las células T humanas e induce una rápida producción de citocinas y reduce la quimiotaxis mediada por CXCR3 de los linfocitos latentes en sangre periférica) y seis meses después había experimentado una remisión completa de la úlcera que se mantuvo todo el tiempo de seguimiento (3 años).
- •
El PG es una dermatosis neutrofílica, que se caracteriza por presentarse como una úlcera necrótica, dolorosa y de rápido crecimiento, con bordes inflamatorios y socavados.
- •
Su etiología y patogenia son desconocidas.
- •
Existen cuatro formas clínicas principales: la ulcerativa o clásica, la pustulosa, la ampollar y la superficial.
- •
El fenómeno de patergia se presenta en un 25% de los casos, por este motivo estos pacientes tienen que evitar traumatismos e intervenciones quirúrgicas innecesarias.
- •
En un el 50% de los casos presenta alguna enfermedad asociada, siendo las más frecuentes la enfermedad inflamatoria intestinal, las artritis y las neoplasias hematológicas.
- •
El ampollar puede presentar manifestaciones extracutáneas, siendo los infiltrados pulmonares estériles la forma de presentación más frecuente.
- •
Es importante realizar una biopsia ante la sospecha de PG. El principal objetivo de esta es descartar otras posibles etiologías.
- •
El diagnóstico del PG se basa en la exclusión de otras posibles causas de úlceras.
- •
No existen estudios controlados que evalúen la eficacia de los tratamientos. Estos se basan en experiencias y recomendaciones de expertos.
- •
Los corticosteroides se consideran el tratamiento de elección, seguido de otros inmunosupresores, e inmunomoduladores así como inhibidores del factor de necrosis tumoral alfa.