





La amenorrea es un motivo de consulta frecuente en atención primaria. El síndrome de Mayer-Rokitansky-Küster-Hauser es una rara anomalía congénita del tracto genital. Se desconoce su etiología. Generalmente se presenta como amenorrea primaria en mujeres adolescentes, con genitales externos y crecimiento normales. Puede asociar otras alteraciones, especialmente a nivel genitourinario. Es preciso realizar pruebas de imagen para confirmar el diagnóstico. No existe consenso en cuanto al tratamiento más adecuado. Exige un manejo multidisciplinar y requiere un correcto asesoramiento y apoyo psicológicos, dado el importante impacto que causa en la esfera psicosocial, tanto de la paciente como de sus familiares1,2.
Exposición del caso clínicoPresentamos el caso de una mujer de 16 años, que consultó inicialmente en atención primaria por no haber presentado nunca menstruaciones. La paciente presentaba un fenotipo femenino. Como antecedentes personales presentaba un astrocitoma difuso (grado ii) frontal izquierdo en seguimiento por neurocirugía; se había desestimado cirugía dada la estabilidad de dicha lesión. La exploración física revelaba caracteres sexuales secundarios femeninos normales (estadio 4 de Tanner), con genitales externos también normales. El resto de la exploración era normal. En la anamnesis, el desarrollo puberal había sido normal. Inicialmente se solicitó analítica con perfil hormonal (prolactina, hormonas tiroideas, FSH, LH, estradiol, testosterona, 17-OH progesterona y DHEA-S), que fue normal. Se derivó al servicio de ginecología, donde se realizó una ecografía abdominal (no se realizó ecografía transvaginal porque la paciente no había tenido relaciones sexuales completas) que fue poco valorable. A pesar de que las determinaciones hormonales fueron normales, se diagnosticó de probable amenorrea central. Con dicha sospecha, fue derivada al servicio de endocrinología, que incidió en dicha sospecha etiológica, atribuyéndose la normalidad de los resultados a error de laboratorio. Se pautó progesterona y, posteriormente, el servicio de ginecología lo sustituyó por una asociación de etinilestradiol y drospirenona. Analíticamente no presentaba hallazgos significativos; el test de LHRH era normal.
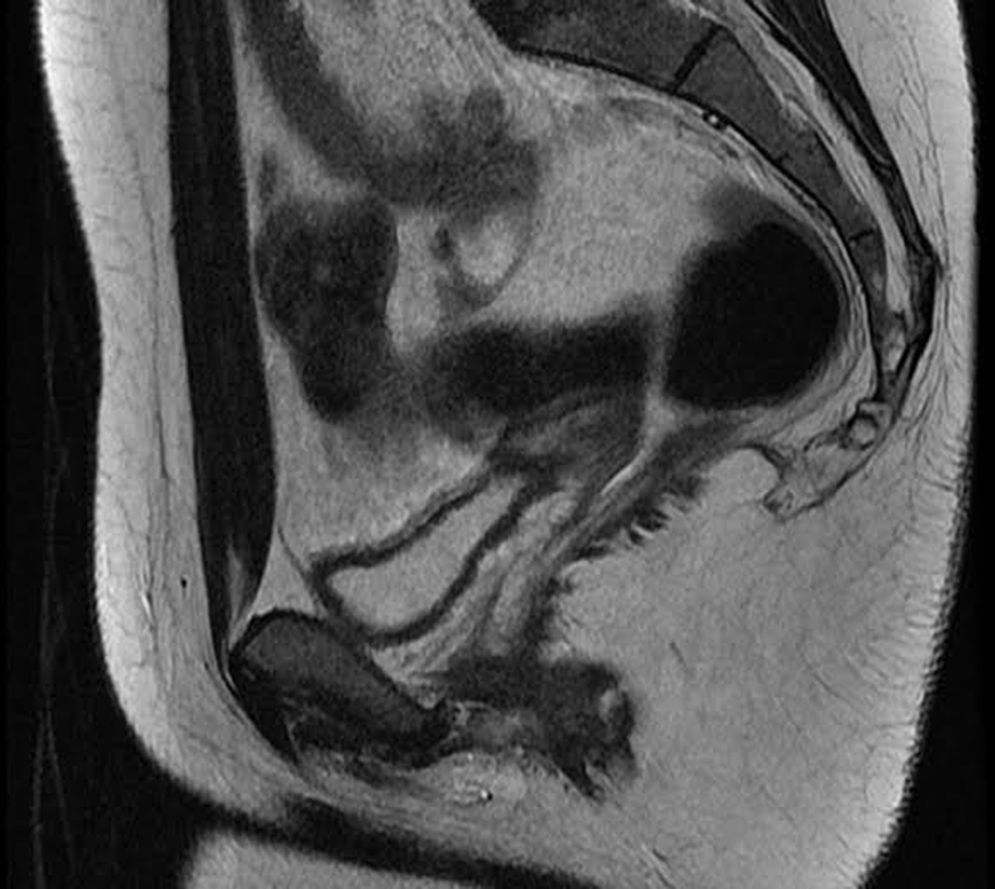
A pesar del tratamiento hormonal pautado por ginecología, 6 meses después persistía la amenorrea, por lo que fue valorada de nuevo en dicho servicio. Se incidió en la exploración ginecológica, apreciándose genitales externos macroscópicamente normales. Ante la imposibilidad para la realización del tacto vaginal y la resistencia al paso de la torunda utilizada en la exploración, se realizó una nueva ecografía donde no se visualizaba útero con certeza, por lo que se le solicitó una resonancia magnética nuclear (RMN) pélvica, en la que se visualizaban ovarios, pero no útero ni cavidad vaginal (fig. 1). Asimismo se solicitó la edad ósea, que era acorde con la edad cronológica. Todo ello era compatible con probable síndrome de Rokitansky. Con dicho diagnóstico fue derivada a servicio de cirugía plástica para valoración de posible tratamiento quirúrgico.

En atención primaria, se indagó más a fondo sobre los antecedentes familiares de la paciente. Tenía una prima por línea materna de 45 años que estaba diagnosticada de síndrome de Mayer-Rokitansky-Küster-Hauser. Había sido intervenida quirúrgicamente mediante laparoscopia, creándose una neovagina mediante la técnica de Vecchietti.
Finalmente, la paciente fue intervenida quirúrgicamente, también mediante la técnica de Vecchietti, creándole una neovagina con evolución posterior satisfactoria.
Desde el momento del diagnóstico hubo que abordar en atención primaria el probable impacto psicosocial que el diagnóstico del síndrome podría conllevar para la paciente y su entorno. Fue necesario apoyo psicológico, farmacológico y orientación, tanto para la paciente como para su familia.
DiscusiónLa amenorrea es un motivo frecuente de consulta en atención primaria. Las causas más frecuentes de amenorrea se describen en la tabla 1.
Causas más frecuentes de amenorrea
| Alteraciones anatómicas |
| - Agenesia mülleriana (síndrome de Mayer-Rokitansky-Küster-Hauser) |
| - Feminización testicular (síndrome de Morris) |
| - Imperforación de himen |
| - Septo vaginal transverso |
| - Síndrome de insensibilidad a andrógenos |
| - Sinequias intrauterinas (síndrome de Asherman) |
| - Agenesia vaginal/cervical |
| - Estenosis cervical |
| Alteraciones eje hipotálamo-hipófisis-ovario |
| Hipotálamo |
| - Causa funcional (estrés, ejercicio, nutricional, enfermedades crónicas) |
| - Infecciones (tuberculosis, lues, meningitis, encefalitis) |
| - Enfermedades inflamatorias/infiltrativas |
| - Radiación craneal |
| - Otros síndromes (síndrome de Prader-Willi, síndrome de Laurence-Moon-Biedl) |
| - Tumores |
| Hipófisis |
| - Tumores hipofisarios. Otros tumores (meningioma, glioma, germinoma) |
| - Síndrome de la silla turca vacía |
| - Infarto/apoplejía hipofisaria |
| Ovario |
| - Disgenesia gonadal |
| - Agenesia gonadal |
| - Fallo ovárico precoz (idiopático, yatrogénico, autoinmune, genético) |
| Otros |
| - Gestación |
| - Enfermedad adrenal (hiperplasia adrenal, enfermedad de Cushing) |
| - Hipo/hipertiroidismo |
| - Diabetes mellitus |
| - Aporte exógeno de andrógenos |
| - Síndrome ovario poliquístico (causa multifactorial) |
Fuente: elaboración propia.
El síndrome de Mayer-Rokitansky-Küster-Hauser (también conocido como aplasia mülleriana) es una rara anomalía congénita del tracto genital. Su incidencia estimada es de una de cada 5.000 mujeres1,2. Se sabe que las alteraciones son el resultado de un desarrollo anómalo (agenesia o hipoplasia) del conducto mülleriano, pero la etiología subyacente es desconocida3–5. Durante años fue considerado una alteración esporádica. A día de hoy, la creciente comunicación de casos familiares hace pensar que pueda tener una base genética. Esta tampoco ha podido ser aclarada de manera concluyente. Parece tratarse de un trastorno multifactorial. En estos casos, se cree que sigue un patrón de transmisión autosómico dominante, con penetrancia incompleta y expresividad variable. Esto sugiere la implicación de mutaciones génicas y/o desequilibrios cromosómicos. Está descrito que el riesgo de recurrencia en parientes de primer grado es bajo, pero existe, fluctuando entre un 1-5%. Típicamente se manifiesta como amenorrea primaria en una paciente adolescente, generalmente alrededor de los 15-17 años. Estas mujeres presentan un cariotipo 46XX. Su desarrollo puberal (salvo por la ausencia de menstruación) y sus genitales externos, son normales1–6. Las pacientes presentan agenesia vaginal con desarrollo uterino variable, pudiendo hallarse un útero rudimentario o ausencia completa de este. Tanto los ovarios como su funcionalidad son normales1–6. En un bajo porcentaje de estas mujeres (2-7%) se aprecia un útero (rudimentario u obstruido) con endometrio funcionante4. Aproximadamente el 25-50% de las pacientes asocian anomalías del tracto genitourinario, tales como agenesia renal unilateral, riñón pélvico o alteraciones del sistema excretor2–7. Asimismo, un 10-15% presentan alteraciones esqueléticas a diferentes niveles (costillas, columna y extremidades). Otras alteraciones asociadas menos frecuentes incluyen cardiopatías congénitas, sordera o hernias inguinales y/o femorales2,5. En el caso de nuestra paciente, coexistían el citado síndrome con un astrocitoma. No hemos logrado encontrar bibliografía en la que se demuestre una relación entre ambos procesos, por lo que podría tratarse de un hallazgo puramente incidental.
Se requiere un alto grado de sospecha para llegar al diagnóstico precoz de esta enfermedad. Las pacientes diagnosticadas antes de la pubertad, generalmente son diagnosticadas de manera incidental, tras estudio por otros problemas de salud al nacimiento o en la infancia. Por otra parte, el diagnóstico puede verse retrasado por la presencia de tabúes sociales.
La valoración de estas pacientes exige la realización de pruebas de imagen2,4,7,8. La ecografía debe realizarse para confirmar la ausencia de útero y la presencia de ovarios, así como para la evaluación de los riñones. Sin embargo, tiene una fiabilidad limitada. Está enormemente influenciada por la experiencia del radiólogo que la lleva a cabo (es operador-dependiente) y, por otra parte, está descrito que cuando dicha prueba se realiza en varias ocasiones puede dar lugar a confusiones. Por ello, si existe dificultad para la visualización de las citadas estructuras, se debería realizar otra prueba de imagen más reveladora, como es la RMN. Esta es útil para valorar la funcionalidad del endometrio en los casos en que se halle útero. El diagnóstico diferencial incluye la insensibilidad a andrógenos, la presencia de un septo vaginal transverso, la agenesia vaginal distal aislada y el himen imperforado2,3,7,8.
Actualmente no existe un consenso en cuanto al tratamiento ideal de estas pacientes. Este debería ir encaminado a la creación de una vagina funcionante. Antes de acometer cualquier terapia debemos asegurarnos de que la paciente ha madurado psicológicamente, pudiéndose requerir para ello una valoración psiquiátrica/psicológica. El tratamiento no quirúrgico con dilatadores presenta un alto índice de éxito, tanto a nivel funcional como por la satisfacción de las pacientes. La cirugía puede estar indicada en casos en que la terapia no quirúrgica haya fracasado, o cuando la paciente la prefiera como tratamiento inicial. La técnica más utilizada por los ginecólogos es la vaginoplastia de McIndoe3. Otros procedimientos menos invasivos, como la técnica de Vecchietti, que se realiza vía laparoscópica, han mostrado últimamente resultados prometedores. Sin embargo, la mejor opción quirúrgica sigue dependiendo de la experiencia del equipo que la realiza y de las preferencias de la paciente. Después de la creación de la neovagina las pacientes pueden llevar una vida sexual normal. Sin embargo, hemos de tener en cuenta que el lograr una vagina anatómicamente intacta, por sí solo, no asegura el éxito del tratamiento. El diagnóstico genera un importante impacto en la esfera psicosocial, tanto en la paciente como en los familiares, por lo que se debe complementar el manejo con asesoramiento y apoyo psicológico, antes incluso de tomar la decisión de iniciar cualquiera de las terapias descritas1,2. Las pacientes afectas por este síndrome presentan importantes tasas de depresión y ansiedad. Por norma general asocian alteraciones de la autopercepción y de la identidad sexual, y ven enormemente obstaculizado el establecimiento y mantenimiento de relaciones interpersonales9. La infertilidad es uno de los aspectos más difíciles de asimilar1,7,10. Compartir dichos sentimientos con grupos de apoyo se ha mostrado beneficioso1. Las respuestas de la familia son variables y se ha demostrado que el apoyo familiar es beneficioso, por lo que se recomienda incentivarlo1.
Este síndrome exige un manejo multidisciplinar, que implica a multitud de profesionales del ámbito sanitario, que deberán tener siempre en consideración las opiniones y anhelos del paciente, y de sus familiares y allegados más próximos2,7–10.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores manifestamos no tener conflicto alguno de interés que interfiera en la publicación del presente trabajo.
