



Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive disease of unknown etiology characterized by the scarring of the lung parenchyma,1 leading to a progressive worsening of dyspnea and lung function, and associated with a poor prognosis.2
Currently, there are two antifibrotic therapies approved for its treatment: pirfenidone and nintedanib.3 However, they are not curative treatments and may be associated with tolerance problems.1 Pirfenidone demonstrated a relative reduction of patients with forced vital capacity (FVC) decline>10%, and nintedanib was shown to reduce FVC loss in absolute value. Therefore, IPF is of particular clinical and investigative interest at present.4 That is why we have made an observational study with retrospective data collection from patients with interstitial lung disease (ILD) who received treatment with an antifibrotic drug in a tertiary hospital in Madrid. The aims of this study were to investigate the changes in clinical and functional characteristics of patients treated with antifibrotic drugs before and after treatment, to identify the diagnosis that led to antifibrotic treatment, and to describe the frequency of adverse reactions to antifibrotic drugs. This study was approved by the Drug Research Ethics Committee of the Gregorio Marañon General University Hospital, with code 13/2023.
The day of antifibrotic treatment initiation was considered baseline of the study. The functional tests included FVC in absolute value and the diffusing capacity of the lungs for carbon monoxide (DLCO) as a percentage of predicted value. These tests were performed according to the Spanish Society of Pulmonology and Thoracic Surgery (SEPAR) guidelines.5 Dyspnea was measured using the modified British Research Council scale (mMRC). The presence of concomitant diseases was measured using the Charlson index. The same functional variables and dyspnea were collected for the 12 months prior to baseline, 12 months after baseline, and 24 months after baseline, with a window of one month if these data were available.
Twenty-eight patients were included, 25 (89.3%) was treated with pirfenidone and 3 (10.7%) with nintedanib. Of all patients, 18 (64.3%) were male. The mean age was 66.4 years (SD 8.4) with a median body mass index (BMI) of 25.2kg/m2 (IQR 22.1–28.4). Only one patient (3.6%) had active smoking at baseline. The median Charlson index was 3 (IQR 2–4).
Regarding the diagnosis, all patients (100%) underwent high-resolution computed (HRCT). In this, 8 patients (28.6%) had a radiological pattern of usual interstitial pneumonia (UIP), 9 patients (32.1%) had a pattern of probable UIP, 3 patients (10.7%) had a pattern of fibrotic non-specific interstitial pneumonia (NSIP), and 8 patients (28.6%) did not fit into any of the previous radiological patterns.
To obtain a histological diagnosis, one patient (3.6%) underwent transbronchial biopsy, 6 patients (21.4%) underwent cryobiopsy, and 5 (17.9%) underwent surgical biopsy. In the case of transbronchial biopsy and cryobiopsy, 5 patients (41.7%) had a histological pattern of UIP, 4 patients (33.3%) had a pattern of NSIP, and 3 (25.0%) showed a histological pattern different from the previous ones. In the 4 patients with surgical biopsy, 3 (75.0%) demonstrated the presence of a histological pattern of UIP. In summary, 22 patients (78.6%) had an established diagnosis of idiopathic pulmonary fibrosis (IPF) as an indication for antifibrotic treatment, and this indication in the other 6 patients (21.4%) was determined by the multidisciplinary ILD committee, made up of pulmonologists, rheumatologists, radiologists and pathologists.
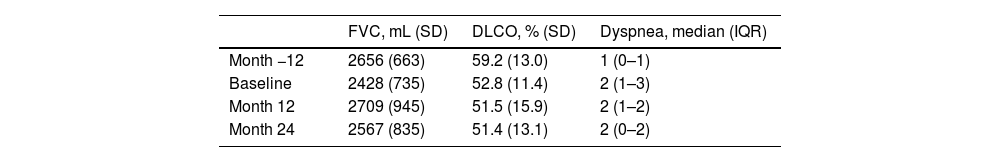
The mean FVC mean at baseline was 2428mL (SD 735), and the mean DLCO was 52.8% (SD 11.4). The comparison of change in FVC, DLCO, and dyspnea between the periods of month −12 to baseline, baseline to month 12, and month 12 to month 24 are presented in Table 1. A lower loss of FVC and less worsening of dyspnea were observed in the 12 months following the initiation of antifibrotic treatment compared to the 12 months prior, without a documented reduction in DLCO during these periods or during the second year of treatment compared to the first year in any of the three variables. The median follow-up was 40.1 months (IQR 32.9–67.1). During the follow-up, 11 patients (39.3%) died, and 2 (7.1%) underwent lung transplantation. In the deceased patients, the median survival from the initiation of antifibrotic treatment was 37.8 months (IQR 33.4–67.1).
Clinical and functional characteristics at month −12, baseline, month 12, and month 24.
| FVC, mL (SD) | DLCO, % (SD) | Dyspnea, median (IQR) | |
|---|---|---|---|
| Month −12 | 2656 (663) | 59.2 (13.0) | 1 (0–1) |
| Baseline | 2428 (735) | 52.8 (11.4) | 2 (1–3) |
| Month 12 | 2709 (945) | 51.5 (15.9) | 2 (1–2) |
| Month 24 | 2567 (835) | 51.4 (13.1) | 2 (0–2) |
FVC: forced vital capacity; DLCO: carbon monoxide diffusing capacity; SD: standard deviation; IQR: interquartile range.
During follow-up, 4 patients (14.3%) experienced gastrointestinal disturbances, 4 patients (14.3%) suffered from photosensitivity, and another 4 (14.3%) had weight loss. These adverse reactions led to temporary suspension of antifibrotic treatment in 5 patients (17.9%) and permanent suspension in 2 (7.1%).
The main finding of our study is that antifibrotic drugs were able to slow down the decline in FVC and worsening of dyspnea during the first year of treatment compared to the previous year, with these parameters stabilizing during the second year of treatment. Results comparable to those found in clinical trials6 and also reported by other authors in a multicenter observational study.7 However, a reduction in the decline of DLCO was not observed with antifibrotic treatment.
Furthermore, most of the patients who received antifibrotics in our study had a confirmed diagnosis of IPF, but in some cases antifibrotic treatment was prescribed by a multidisciplinary ILD committee even when the patient did not have IPF. In our study, 64.3% of the patients were male. As previously described in the literature, the incidence and prevalence of IPF are higher in males, and the disease progresses more rapidly in this population.8,9 Radiology plays an essential role in the diagnosis of IPF, and it can even be diagnostic without the need for anatomopathological confirmation,10 as was the case in 16 of our patients.
Antifibrotic drugs were safe and well tolerated by the majority of patients, with temporary suspension or withdrawal necessary in 25% of patients, although no serious adverse effects were detected. These data are comparable to the results of the CAPACITY study11 and are also similar to those found in various real-life observational studies, with gastrointestinal disturbances being the most frequent adverse effects.6,7,12
In conclusion, the antifibrotic treatments slowed the decline of FVC and worsening of dyspnea during the first year of treatment compared to the previous year. These treatments were generally well tolerated. These findings encourage the continued use of antifibrotics in indicated cases.
FundingThis study received no external funding.
Authors’ contributionsConceptualization: Katiuska Herminia Liendo-Martínez
Methodology: Zichen Ji
Software: Zichen Ji
Validation: Francisco Javier Baratech-Calpena
Formal analysis: Zichen Ji
Investigation: Katiuska Herminia Liendo-Martínez and Francisco Javier Baratech-Calpena
Resources: Fernando Pedraza-Serrano and Francisco José Caballero-Segura
Data curation: Katiuska Herminia Liendo-Martínez
Writing—original draft preparation: Katiuska Herminia Liendo-Martínez and Francisco Javier Baratech-Calpena
Writing—review and editing: Javier de Miguel-Díez
Visualization: Javier de Miguel-Díez
Supervision: Javier de Miguel-Díez
Project administration: Javier de Miguel-Díez
Funding acquisition: N/A
Conflicts of interestThe authors declare that they have no conflict of interest directly or indirectly related to the contents of this manuscript.
