



Introducción: El síndrome de hipoventilación alveolar central congénita (SHACC) es un raro trastorno respiratorio del dormir, aunque cada vez más frecuentemente diagnosticado en clínicas de sueño y servicios de neumología pediátrica. Si bien se desconoce su epidemiología, en la literatura médica existen cerca de 300 casos reportados, y su incidencia es de 1 caso por cada 200,000 recién nacidos vivos. Se caracteriza por hipoventilación alveolar que se presenta o empeora durante el sueño. Es secundario a la disminución/ausencia de la respuesta ventilatoria a la hipercapnia o hipoxemia, y en el 90% de los casos es debido a una mutación tipo PARM del gen PHOX2B. Su tratamiento incluye ventilación mecánica y marcapasos diafragmático. Si la terapéutica no se inicia en forma temprana, el paciente desarrollará insuficiencia respiratoria crónica, hipertensión arterial pulmonar, cor pulmonale y la muerte.
Casos clínicos: Se presentan tres casos de SHACC diagnosticados, tratados y en seguimiento en la Clínica de Trastornos Respiratorios del Dormir del Instituto Nacional de Enfermedades Respiratorias.
Conclusiones: El diagnóstico temprano es importante para el inicio del soporte ventilatorio, y para prevenir el desarrollo de complicaciones y reducir la mortalidad.
Background: Congenital central hypoventilation syndrome (CCHS) is a rare sleep-related breathing disorder. Although increasingly frequently diagnosed in sleep clinics and pediatric pulmonology services, its epidemiology is not known. There are about 300 reported cases in the literature with an incidence of 1 case/200,000 live births. CCHS is characterized by alveolar hypoventilation that occurs or worsens during sleep and is secondary to a reduction/absence of the ventilatory response to hypercapnia and/or hypoxemia. In 90% of the cases it is due to a PARM-type mutation of the PHOX2B gene. Treatment includes mechanical ventilation and diaphragmatic pacemaker. If therapy is not initiated promptly, the patient can evolve to chronic respiratory failure, pulmonary hypertension, cor pulmonale and death.
Case reports: In this paper we present three cases of CCHS diagnosed, treated and followed-up at the Sleep Disorders Clinic of the National Institute of Respiratory Diseases in Mexico.
Conclusions: Early diagnosis is important to initiate ventilatory support so as to prevent any complications and to reduce mortality.
1. Introduction
Congenital central hypoventilation syndrome (CCHS) is a rare condition characterized by alveolar hypoventilation that presents itself or worsens during sleep. This hypoventilation is secondary to the decrease or lack of ventilatory response to hypercapnia or hypoxemia. By definition, it should not be a specific consequence of central nervous system, metabolic, pulmonary, cardiac disease or other lesions that explain the hypercapnia1. The disease spectrum varies and although the majority of cases are diagnosed neonatally, cases of late onset have been described. The exact epidemiology is unknown, but in the international literature there are almost 300 reported cases and an incidence of 1/200,000 live newborns have been reported in France2.
We present three cases of children with CCHS treated in the Sleep Respiratory Disorders Clinic of the National Institute of Respiratory Diseases (INER) of México.
2. Clinical Cases
2.1. Case 1
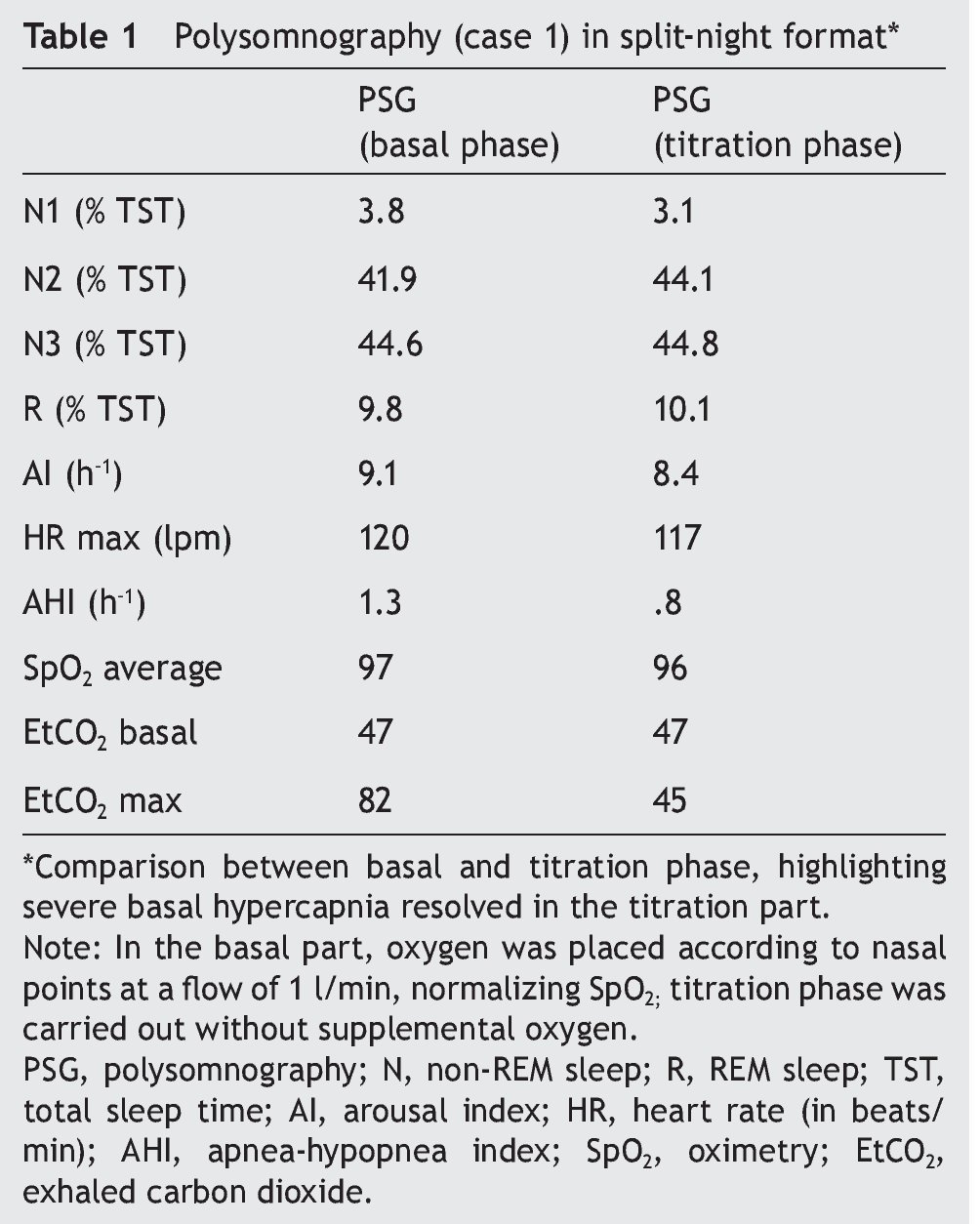
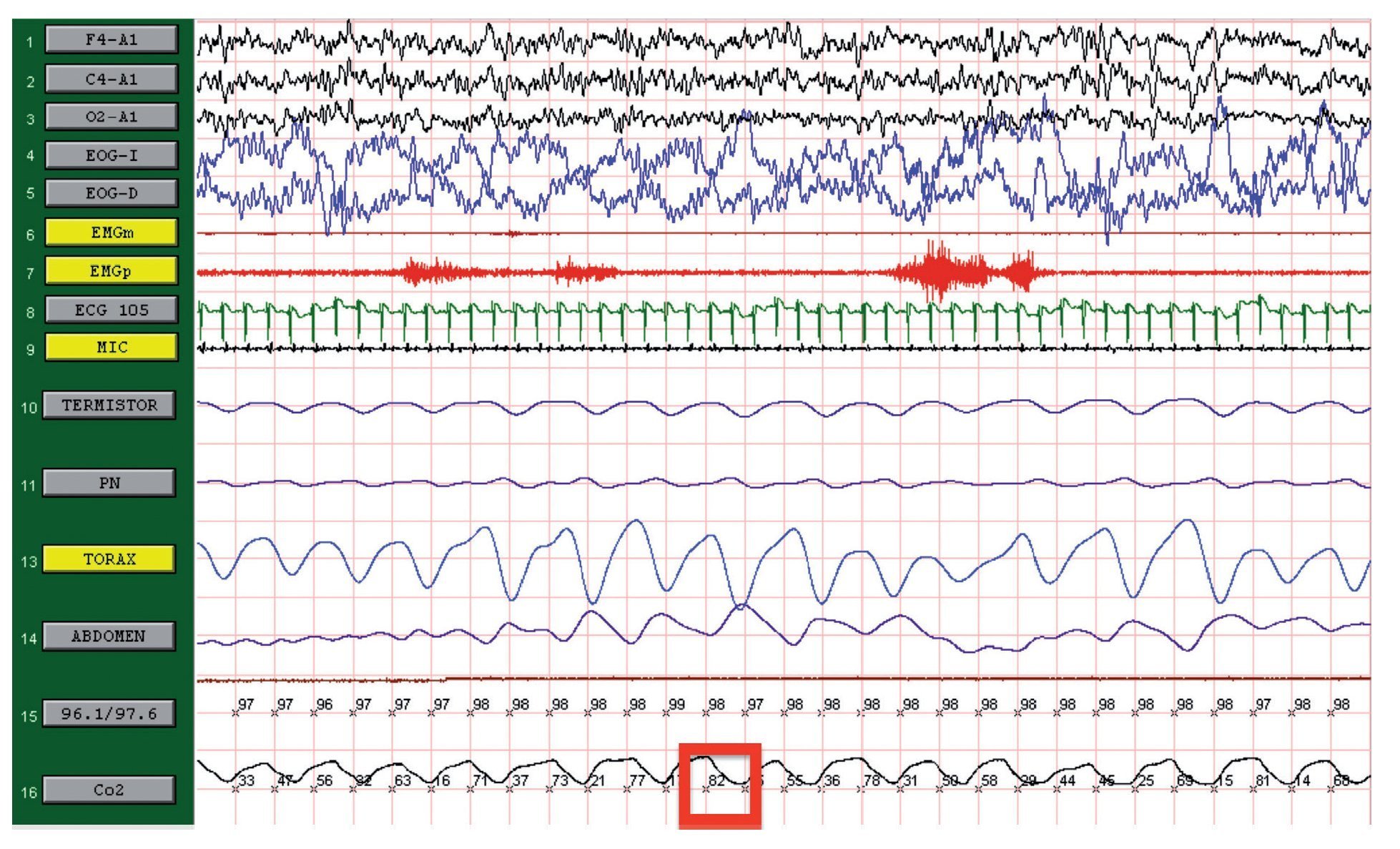
We present the case of a 12-year-old male patient weighing 20.6 kg and measuring 125 cm in height who was hospitalized for community-acquired pneumonia and subsequently sent to this service for chronic hypercapnia and cor pulmonale. He did not manifest symptoms of autonomic dysfunction; his sleep was described as normal, without snoring or apneas. Severe chronic respiratory insufficiency was documented by oximetry of 57%, exhaled carbon dioxide (EtCO2) of 47 mmHg (Table 1), blood gases with pH 7.4, PaCO2 45.4 mmHg, PaO2 44.1 mmHg, HCO3 27.8 mmol/l, SaO2 79.9%. Polysomnography (PSG) found a maximum EtCO2 of 82 mmHg (Figure 1); respiratory function tests and chest x-rays did not show any changes. Electrocardiogram found a right ventricular overload, echocardiogram with enlarged cavities and pulmonary systolic pressure 34 mmHg. The remaining laboratory studies were without changes. Treatment was initiated with noninvasive positive pressure ventilation (NIPPV) and was maintained for 9 months, but the NIPPV was stopped and the patient died 6 months later due to acute respiratory failure and cor pulmonale. A genetic analysis was not performed.
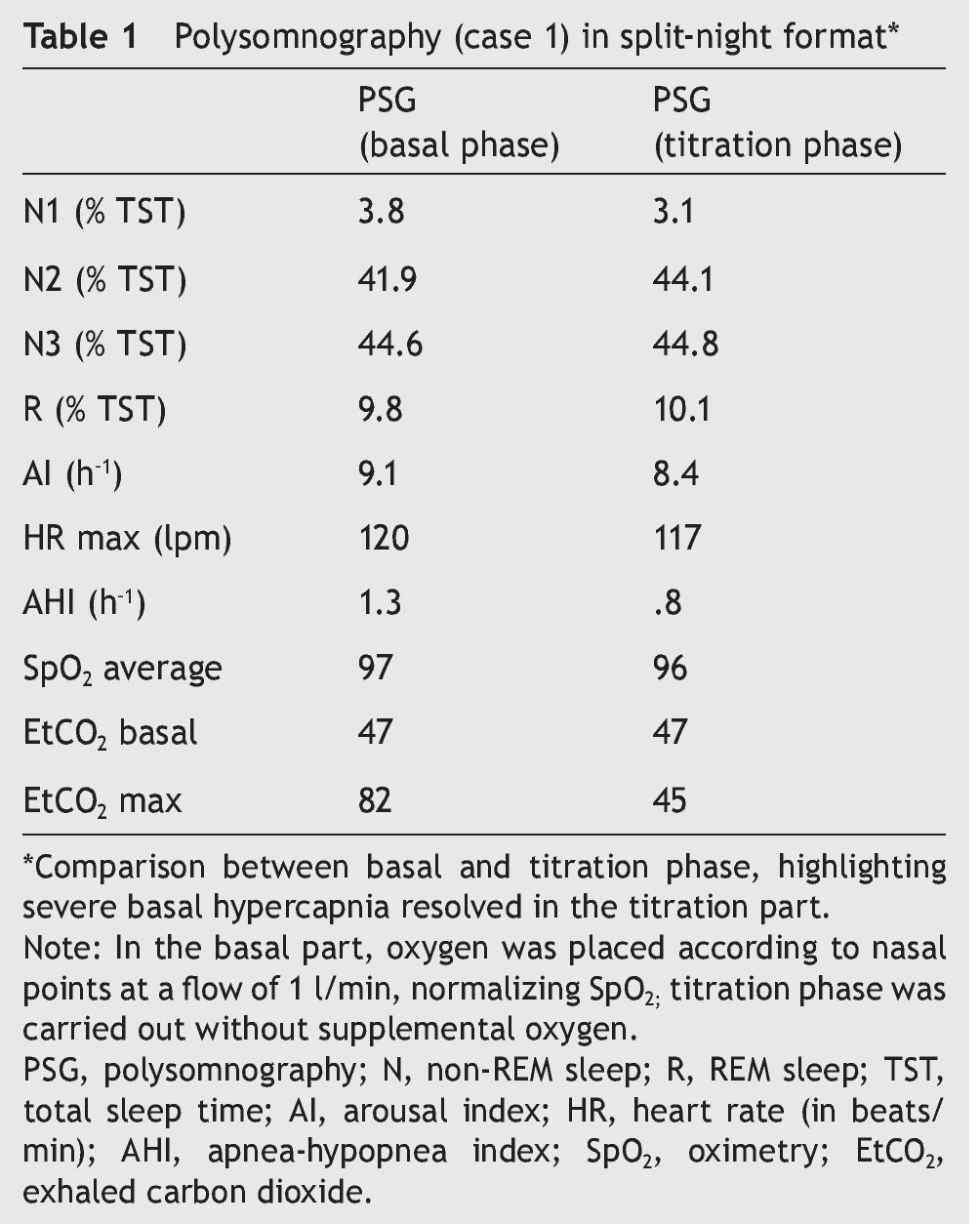
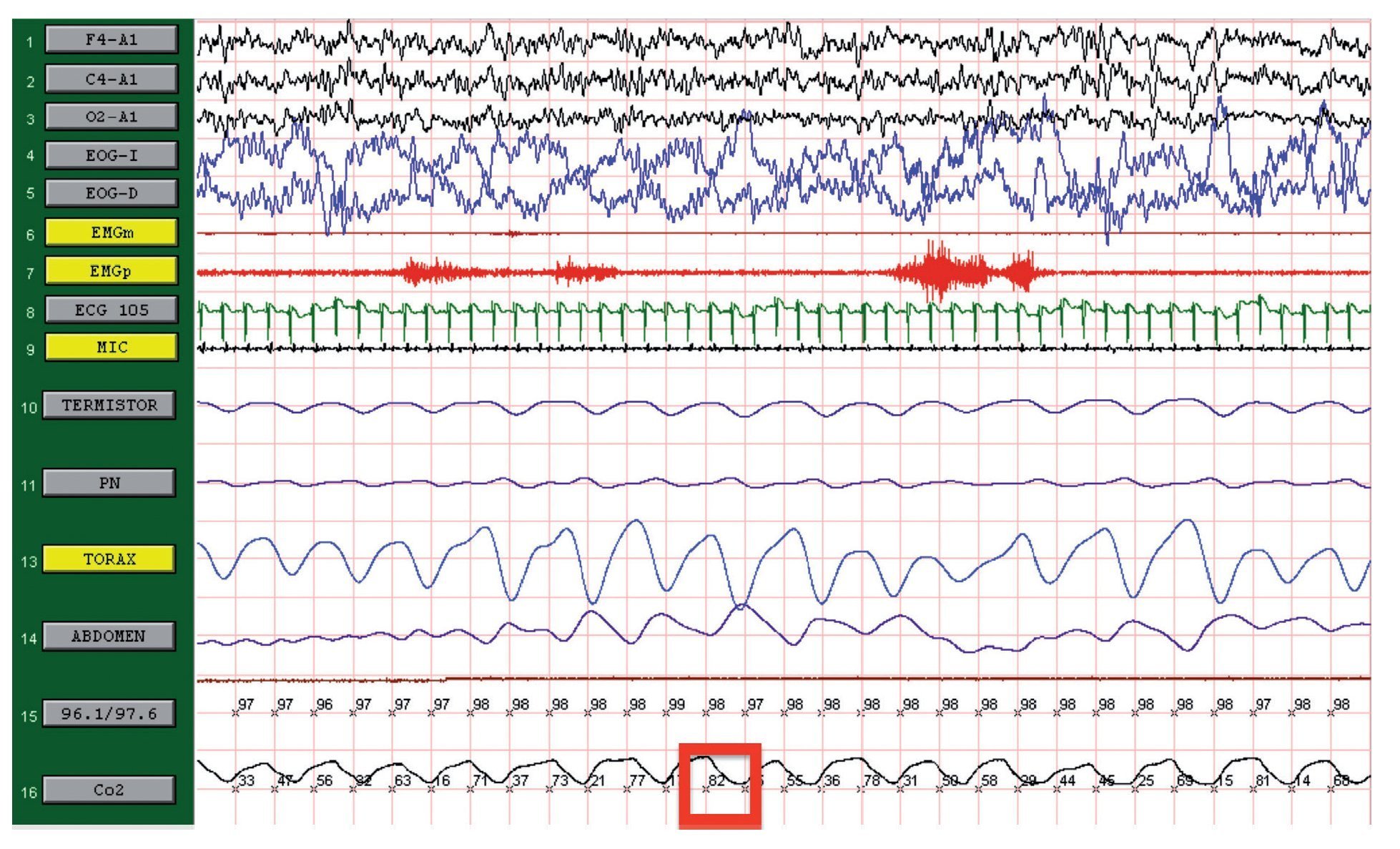
Figure 1. Polysomnography (case 1). The patient is in stage 1 of non-rapid eye movement sleep. Maximum level of EtCO2 (red frame) showing severe hypercapnia during sleep. EtCO2, exhaled carbon dioxide.
2.2. Case 2
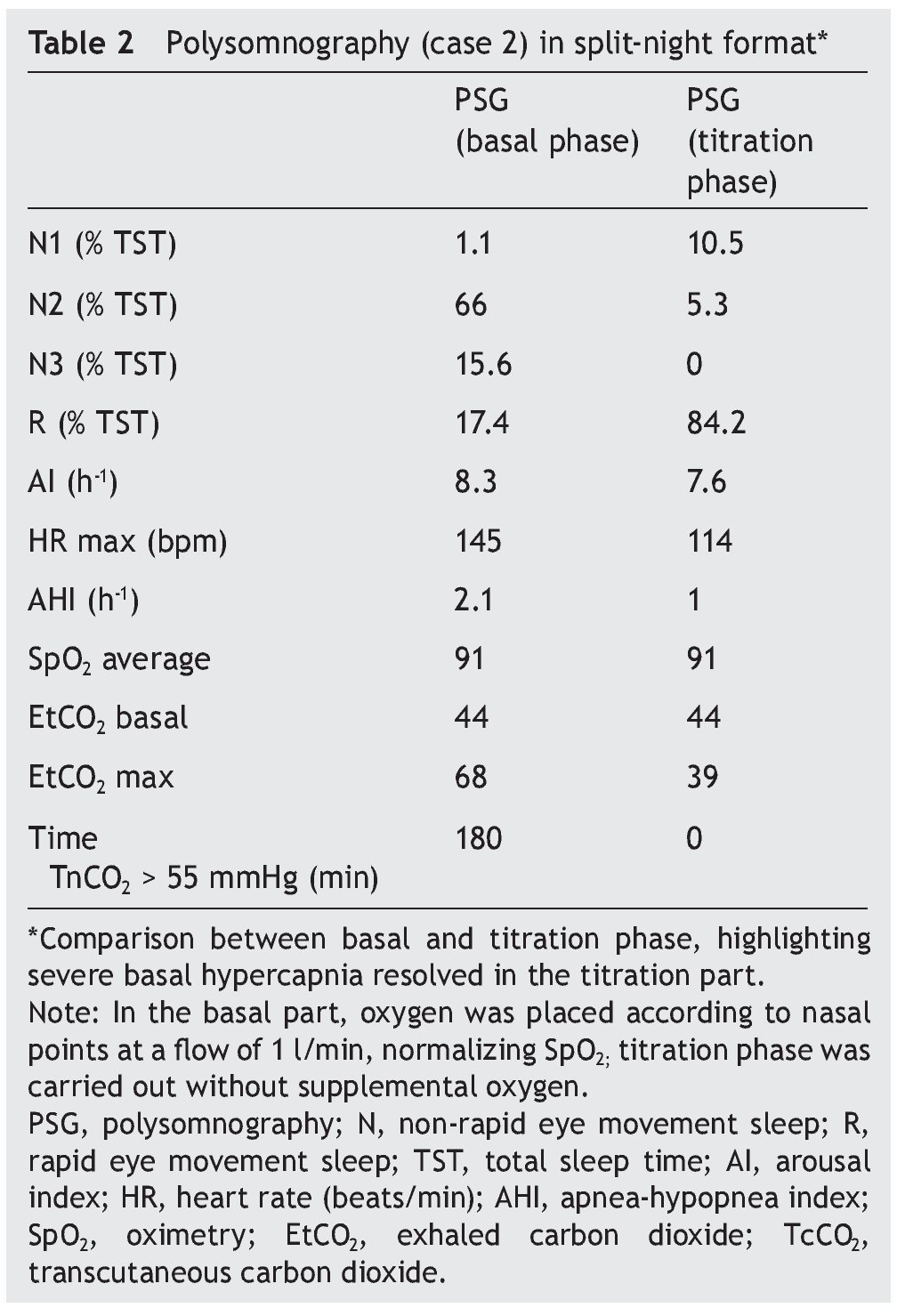
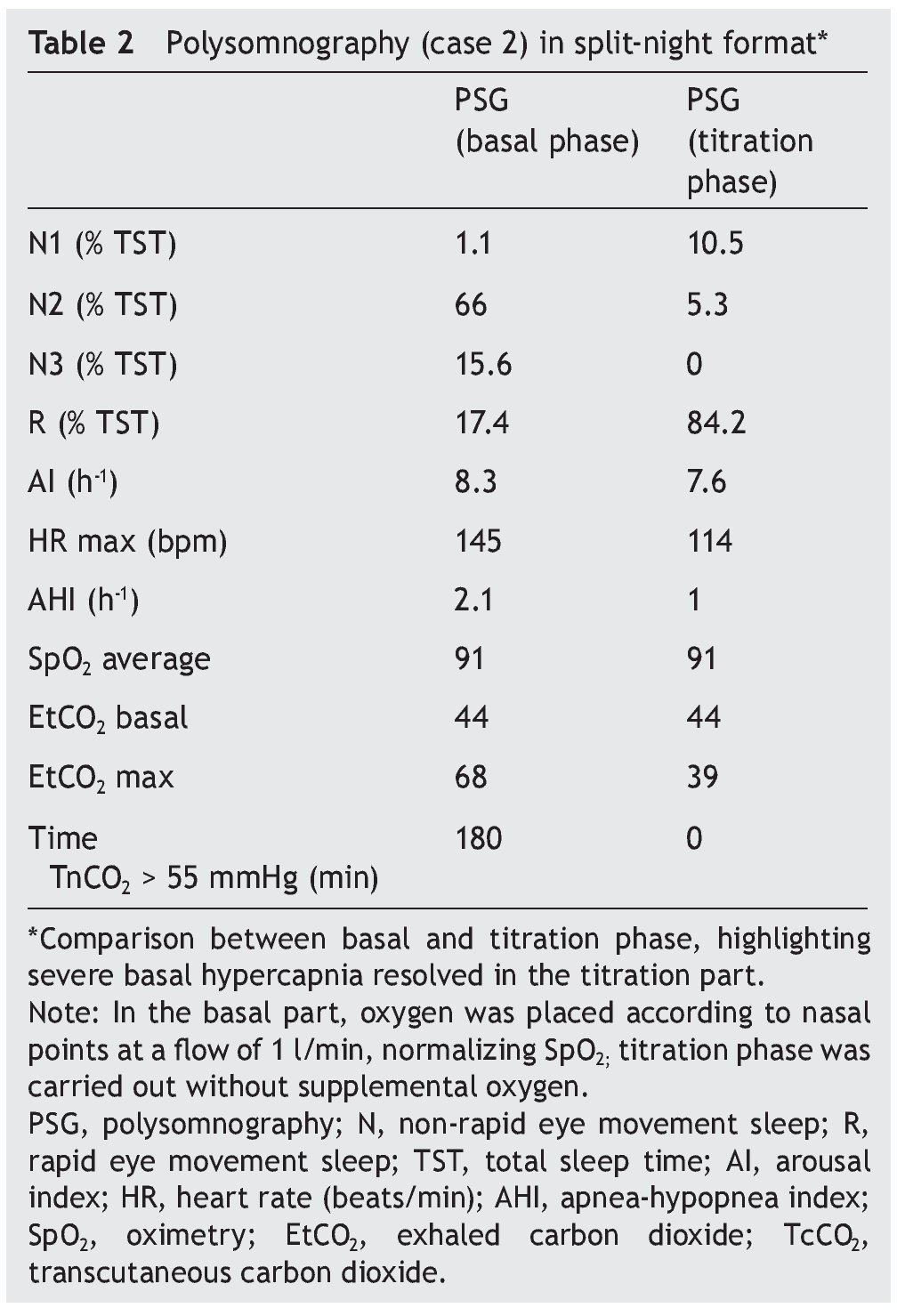
We present the case of a 1 year 3 month-old male patient who was the product of a term pregnancy of normal evolution with age-appropriate growth and development. The patient was hospitalized for community-acquired pneumonia and acute severe respiratory hypercapnia, apparently disproportionate to the symptoms. He developed seizures and exaggerated response to benzodiazepines and required management with invasive mechanical ventilation and subsequent difficulty weaning due to hypercapnia. During his stay in the intensive care unit, nocturnal episodes of tachycardia/bradycardia and diaphoresis were identified. Daytime oximetry was 91%, diurnal EtCO2 was 44 mmHg, nocturnal EtCO2 was 66 mmHg, diurnal PaCO2 was 41 mmHg, and nocturnal PaCO2 was 120 mmHg. Nuclear magnetic resonance (NMR) studies of the brain, EEG and chest x-ray were normal, and echocardiogram showed mild growth of the right cavities. PSG was done and severe nocturnal hypoventilation was documented. A positive pressure device was introduced (Table 2). The patient is currently stable and treated with bi-level NIPPV with respiratory rate support (RRT). Genetic analysis was not carried out.
2.3. Case 3
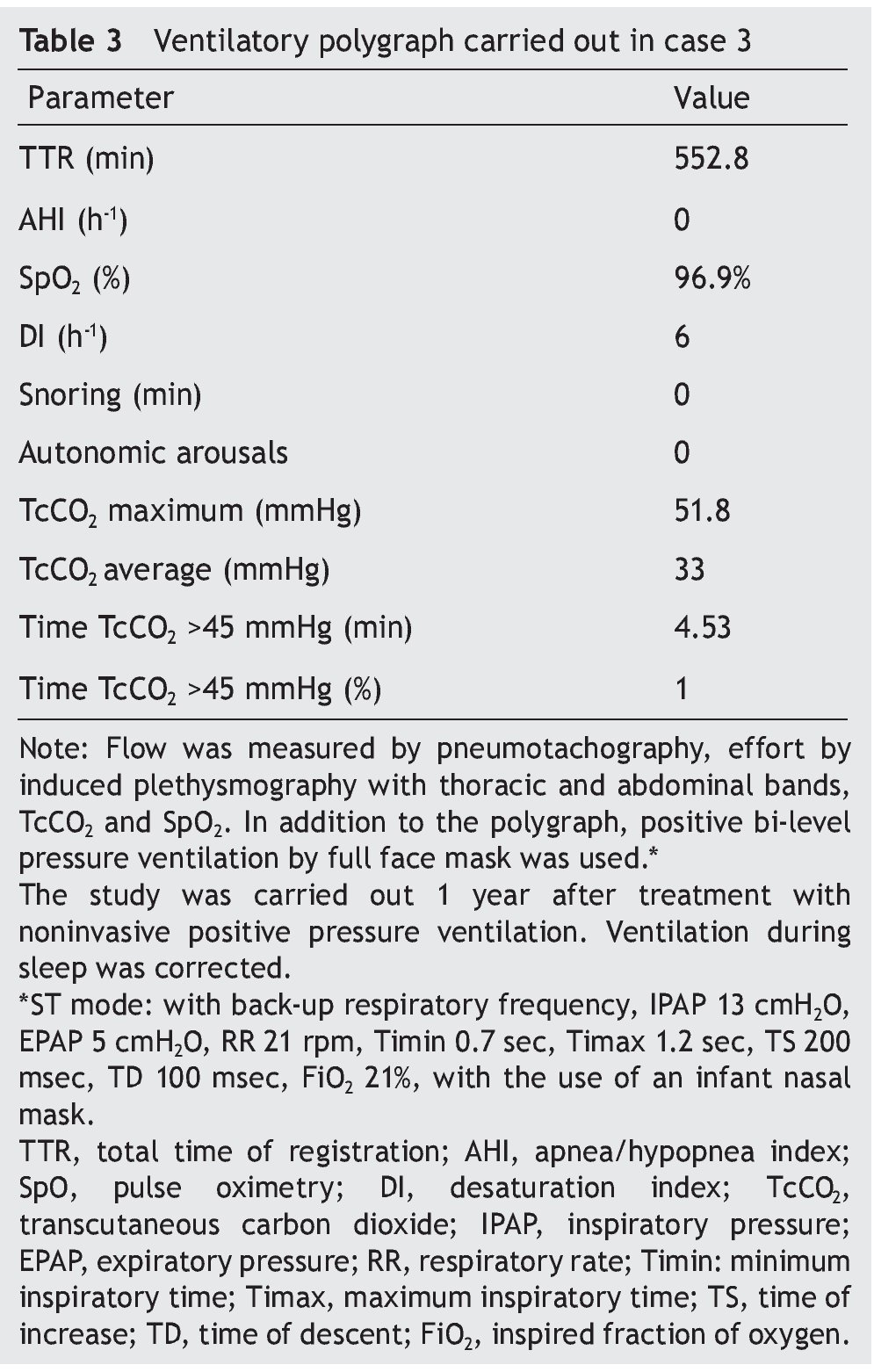
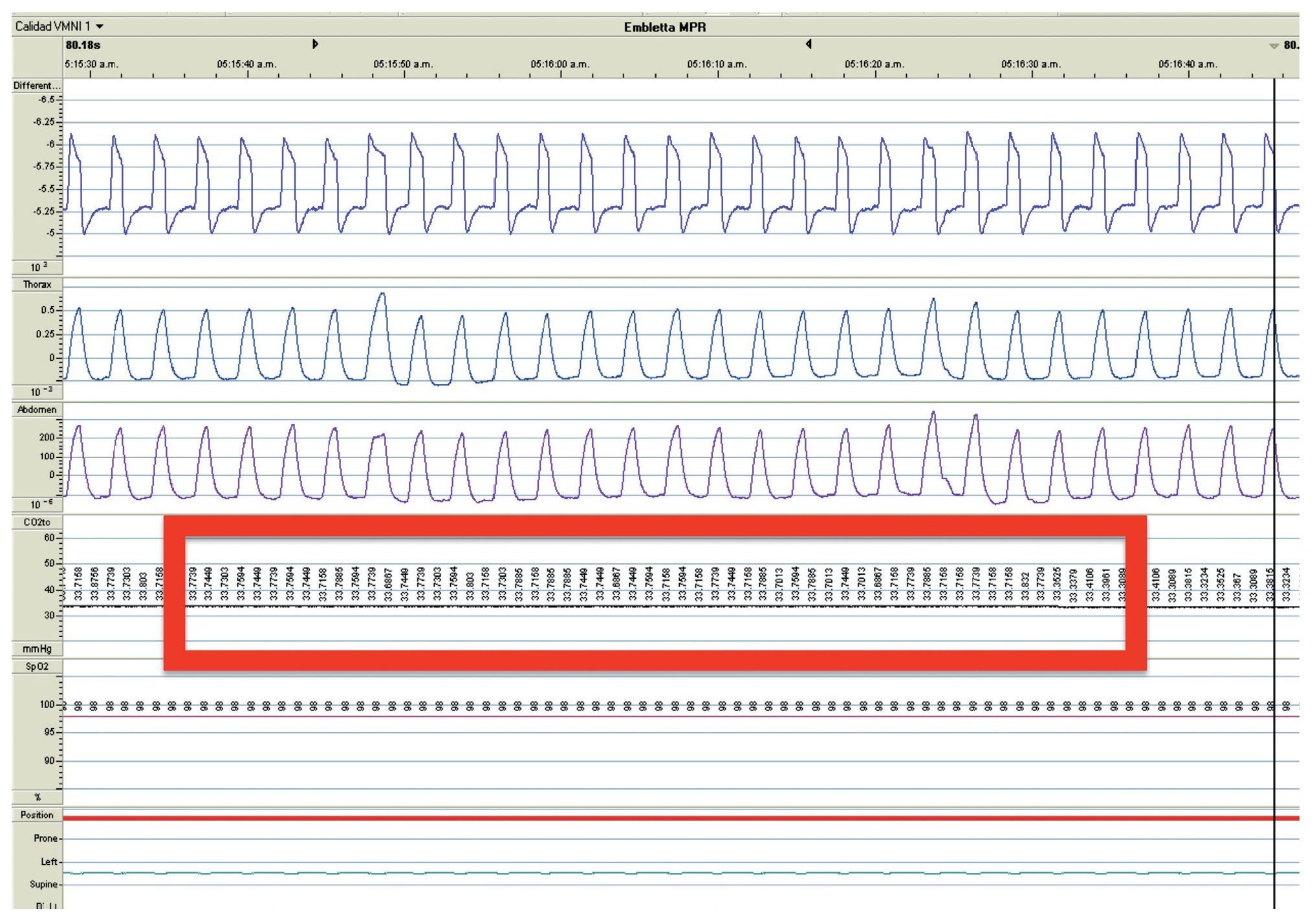
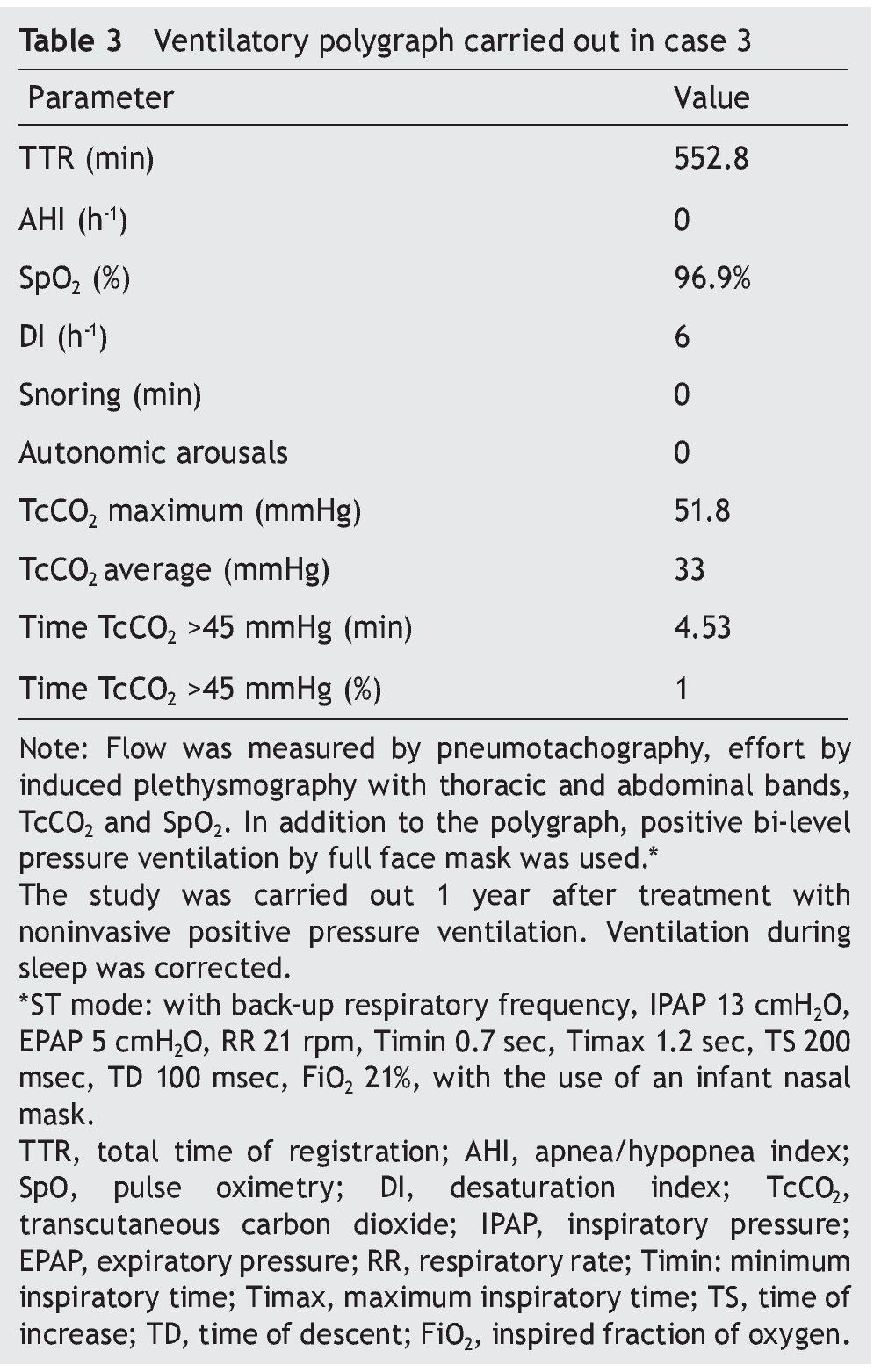
We present the case of a 2 year 8 month-old male patient born by cesarean section at 40 weeks gestation and with an Apgar score of 9/10. He was hospitalized from birth for 3 months due to hypoxia attributed to permeable foramen ovale. This malformation was corrected at 6 months of age; however, the hypoxemia persisted. During the first year of life he presented three episodes of respiratory failure attributed to pneumonia. At 1 year 9 months of age pulmonary arterial hypertension was documented during the study for hypercapnic respiratory failure. During the acute phase of the respiratory failure he developed seizures, for which he required invasive mechanical ventilation for 9 days. During hospitalization, apnea was noted without inspiratory effort during sleep, desaturations and severe hypercapnia with PaCO2 of 110 mmHg. During the initial evaluation, a child with age-appropriate growth and development was observed, diurnal oximetry during wakefulness of 84% and EtCO2 of 64 mmHg. During PSG (Figure 2) the following findings were made: normal sleep architecture, apnea hypopnea index of 3.5 events per hours of sleep, maximum transcutaneous CO2 (TcCO2) of 160 mmHg and hypoxia corrected by low flow oxygen supplementation. Treatment was then begun with a NIPPV device in bi-level ST mode. A study of the PHOX2B gene was requested and was positive for CCHS (genotype 25/20). After 1 year of treatment with a NIPPV bi-level ST mode device, he has not required hospitalization for any reason. Diurnal gas exchange was reported as normal (SpO2 94%, EtCO2 34 mmHg, blood gases with pH 7.4, PaO2 76 mmHg, PaCO2 34 mmHg, HCO3 24 mmol/l, EB-2, SaO2 94%) and compensated nocturnal ventilation (Table 3).
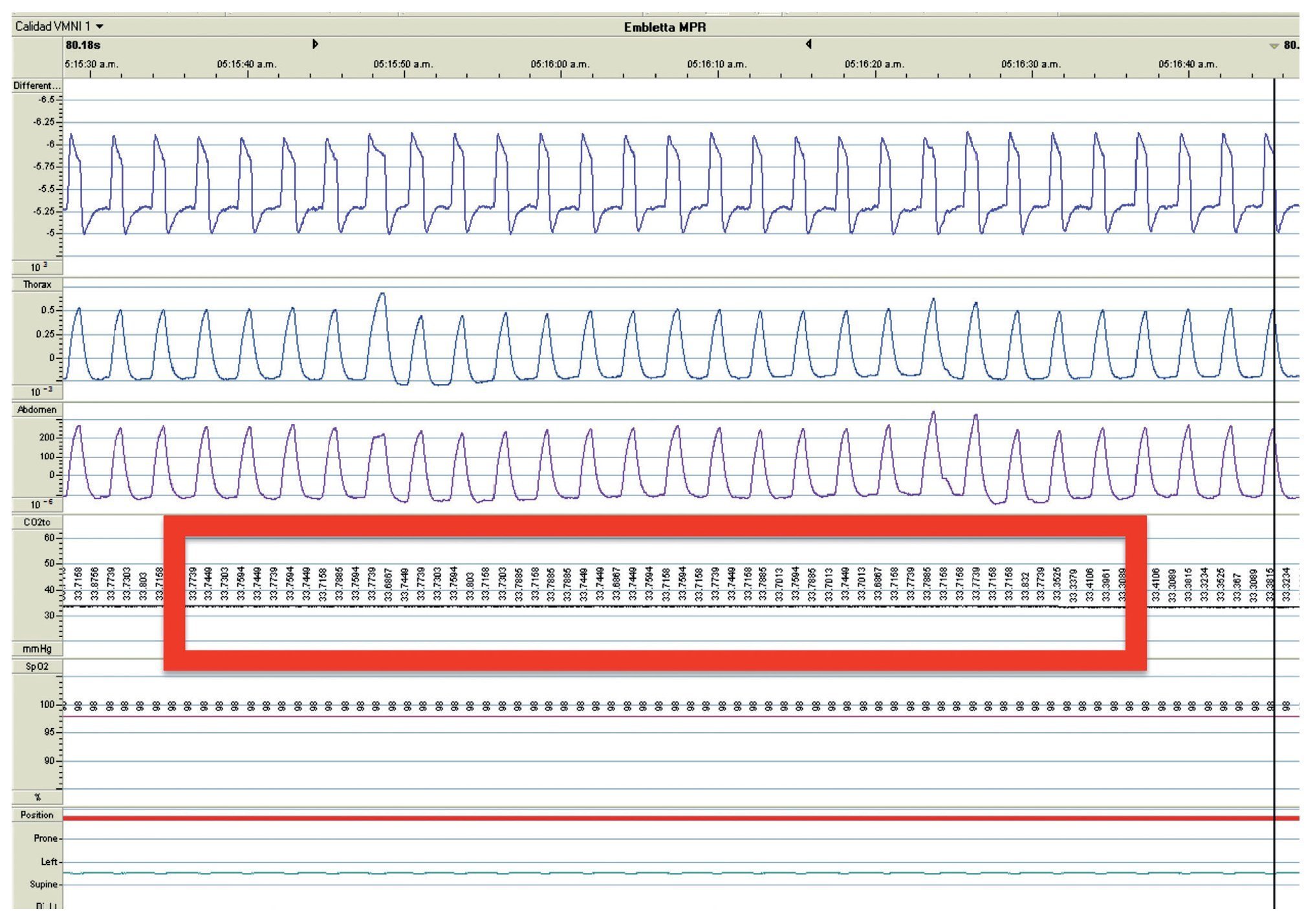
Figure 2. Nocturnal ventilatory polygraphy after 1 year of follow-up of case 3. From top to bottom, the signals shown are flow by pneumotachograph, inspiratory effort by band on chest and abdomen (inductance plethysmography), TcCO2 and SpO2. Highlighted are the TcCO2 values of 33 mmHg (red frame). SpO2, pulse oximetry; TcCO2, transcutaneous carbon dioxide.
3. Discussion
CCHS is a disease of the autonomic nervous system and respiratory control and characterized by a decrease or absence of the ventilatory response to hypercapnia or hypoxemia in the absence of specific organic lesions3-5. It was described for the first time in 1970 by Mellins et al.6. In 2003 it was discovered that the mutations of the PHOX2B gene located on the 4p12 chromosome are responsible for this syndrome7,8.
There are ~300 cases reported in the international literature and, although its incidence in the general population is unknown, in 2005 Trang et al., based on the creation of a registry of these cases in France, estimated an incidence of 1/200,000 live newborns2. The number of cases reported has increased in recent years, especially in the Caucasian population. However, this increase is possibly due to an expanded search and better identification of cases. Studies based on larger populations and different ethnic groups are necessary to determine the true prevalence of this disease5.
The exact pathophysiological mechanism of the disorder in respiratory control is not yet clear. It has been observed that the majority of patients with this disease have an adequate control of ventilation during wakefulness and alveolar hypoventilation secondary to the reduction of the tidal volume during sleep, which is more severe during the stage of non-rapid eye movement sleep. In severe cases hypoventilation has been observed both during wake state as well as during sleep9.
Lack of response of the chemoreceptors has been observed in various studies both to the hypercapnia as well as the hypoxemia, even during the wake state, in children with CCHS; however, the state of alert for hypercapnia is preserved, although attenuated, and the response of the peripheral chemoreceptors appears to be less affected or intact in children with adequate ventilation during wakefulness. These observations support the hypothesis that there is an alteration in the integration of the information received by the chemoreceptors in the respiratory control center3,10-13.
The genetic origin of the disease was postulated based on the concordance of the disease with homozygous twins14,15. In 2003 it was discovered that mutation of the PHOX2B gene is the cause of the disease and has a dominant autosomal inheritance pattern, although in most cases a spontaneous mutation is produced (90-95%)5,16. The PHOX2B gene codes a transcription factor that regulates cellular migration during formation of the neural crest and development of the central nervous system (CNS) (neurocristopathies)7,8,16. This explains the association observed between the severe forms of CCHS and autonomic dysfunctions, Hirschsprung disease and neural crest tumors16,17; 90% of the cases of CCHS are heterozygous to the mutation of repeated expansion of polyalanine (PARM), in the PHOX2B gene with a range from 4-13 additional repetitions (genotypes from 24/20 to 33/20). The number of mutations is associated with the severity of the control disorder19. The remaining 10% has another type of mutation unrelated to polyalanine expansions (NPARM), which are associated with more severe clinical forms of neural crest tumors20.
At the time of the diagnosis the clinical picture can be divided according to the age when it began:
• Neonatal initiation—the average age of diagnosis is 3.5 months2. Patients with CCHS tend to present intermittent episodes of cyanosis or apnea at birth and many require immediate mechanical ventilation after birth but do not tend to have evidence of any other neurological lesion that explains the ventilatory disorder. In another group it presents itself in the first months of life with life-threatening episodes and even respiratory arrest during sleep1,5. In many patients a regular, but superficial respiratory pattern is observed during sleep, with reduction in movement of the chest wall and episodes of central apneas; also, hypercapnia and hypoxemia can be documented that are not associated with an increase in the effort or with the respiratory rate. No signs of alert are seen and the patient tends to appear calm. In many children the hypoventilation can be seen in the awake state, but with eventual growth they will breathe adequately during the awake state due to the maturation of the CNS, growth of the respiratory apparatus (increase in alveolar units and improvement in the ratio of the dead space volume/tidal volume) and decrease in total sleep time1,3,5. These changes can be a false and transient expression of cure; however, the decrease/absence of ventilatory response to hypercapnia/hypoxia persist, for which treatment requirement continues to be necessary.
In case of inadequate diagnosis, these children develop signs of right heart insufficiency and PAH. Hypoventilation tends to worsen, even with mild respiratory infections because children are not able to increase their ventilatory response to increased demands and do not tend to present obvious signs or symptoms of respiratory failure. These patients, in general, have a poor response to exercise or stress21.
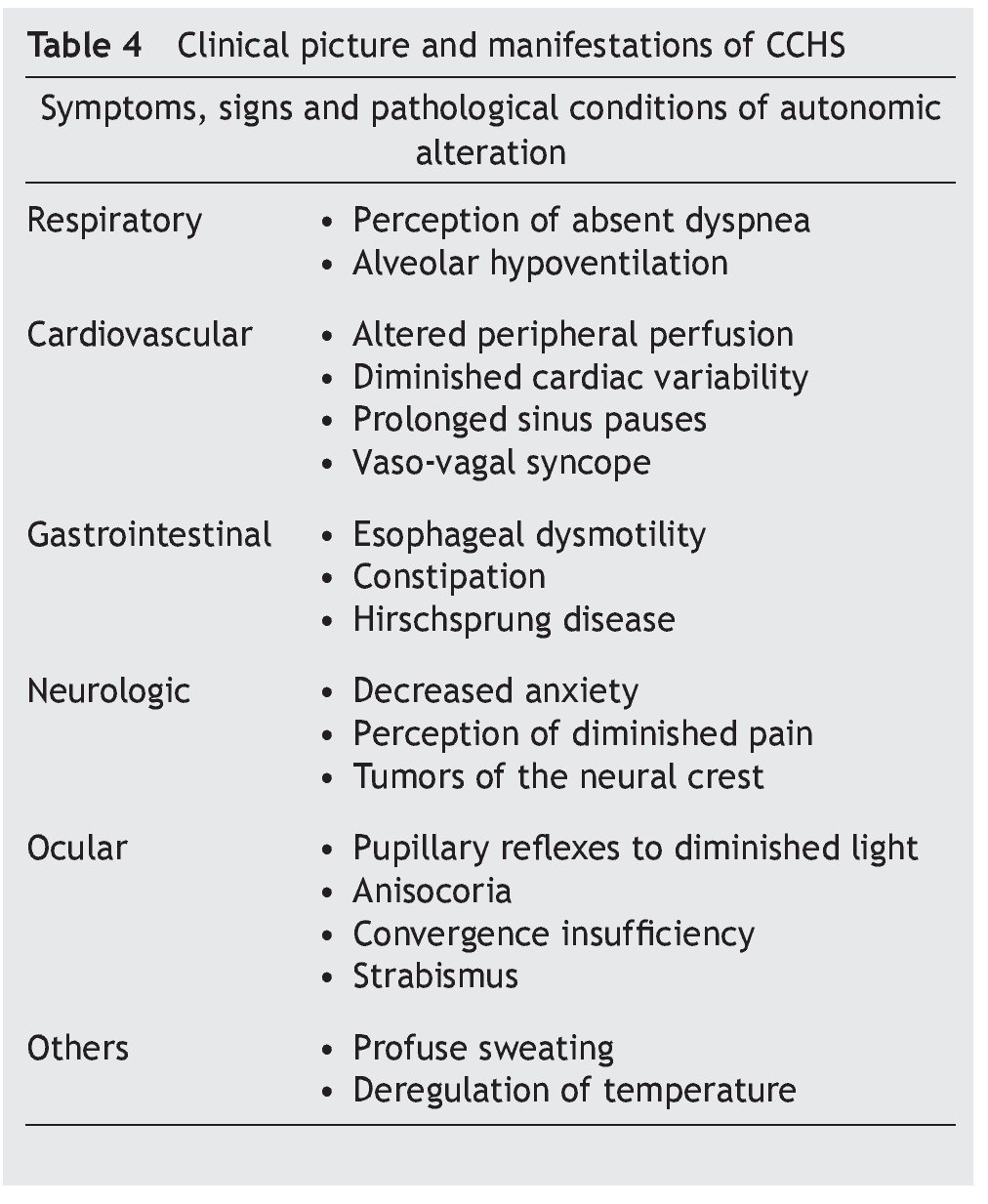
Some infants with CCHS may present signs of target organ damage such as cor pulmonale, seizures and psychomotor delay secondary to chronic hypercapnia and hypoxemia not previously detected1,3,5. In addition, problems with growth and craniofacial dysmorphias have been described. Children with autonomic dysfunction have been found such as diaphoresis, tachycardia/bradycardia, tachypnea/bradycardia, altered pupillary response, and alteration in temperature regulation, among others (Table 4)22-24.
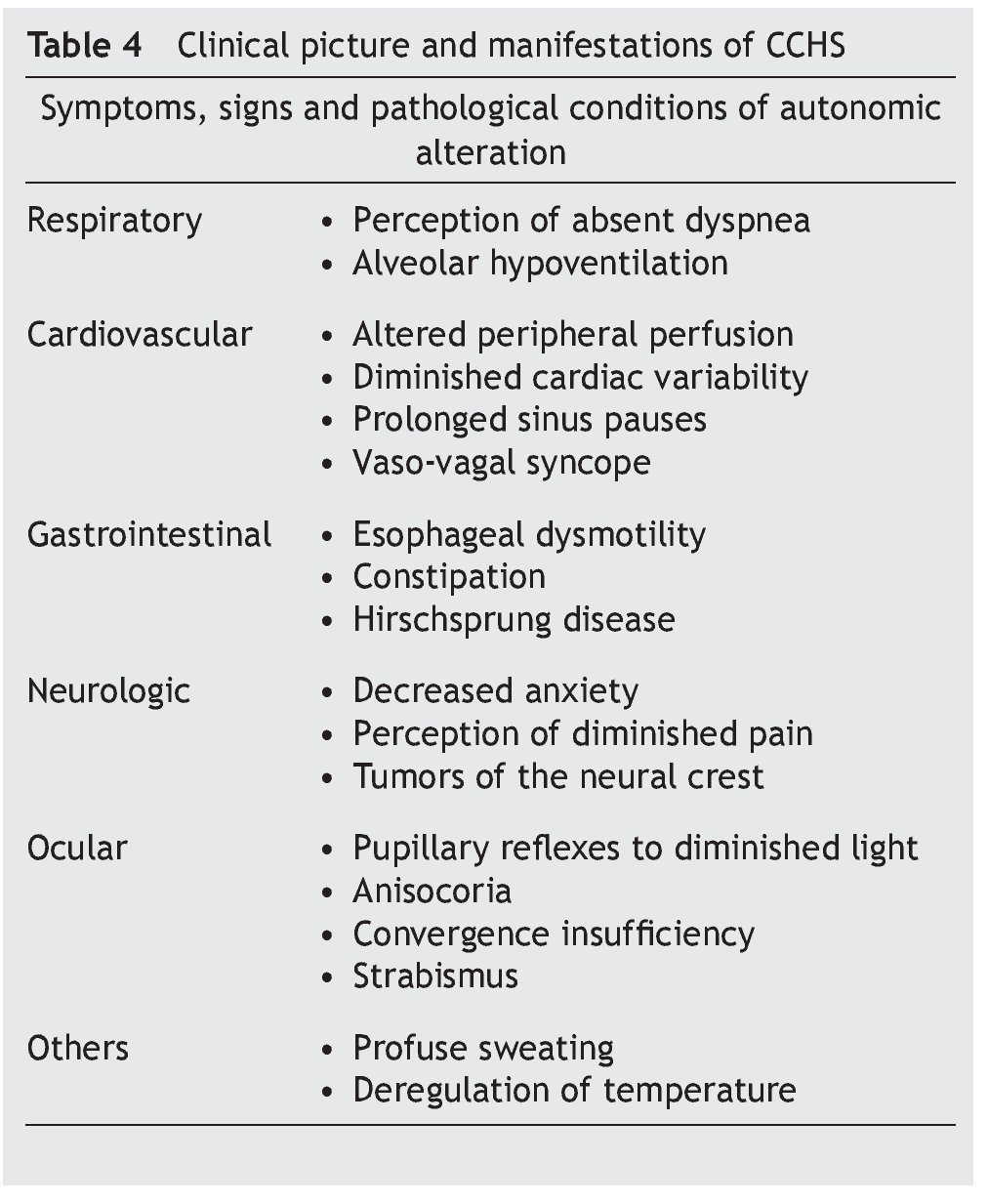
• Late-onset complaints—presentation in older children or in adulthood is still rarer. The severity of the disease depends on the genotype, which is usually associated with short mutations (23/20–25/20), which have a tendency to be observed in patients with some type of neurocognitive disorder, epilepsy, sleep apnea or history of hypoventilation during infancy. Occasionally it can be detected by hypoventilation after general anesthesia or respiratory infections (Table 4)25-29.
Diagnostic criteria proposed by the American Academy of Sleep Medicine were actualized in 2014 (Table 5)30,31.

3.1. Initial Evaluation
• Diagnostic suspicion should be established in those patients with clinical picture suggestive of hypoventilation without obvious organic cause1,4,5, normal physical examination, changes in respiratory pattern and hypoventilation during sleep and signs of chronic hypoxemia and hypercapnia.
3.2. Diagnostic Tests
• Detection of the mutation of the PHOX2B gene should be done in all patients with clinical suspicion; 90% of the cases of CCHS are positive for this gene mutation; however, with a negative test result, a clear clinical pheno-type is sufficient for diagnosis4,5,32.
• Polysomnography (PSG) with capnometry through transcutaneous or continuous exhaled measurement is necessary to study the level of hypoventilation during sleep9,33,34.
• Additional evaluations. Additional exams should be performed to evaluate the severity and chronicity of the disease as well as to identify the associated autonomic changes1,4.
Erythrocytosis: hematocrit and hemoglobin
Chronic respiratory acidosis: arterial blood gases, serum bicarbonate
Pulmonary hypertension–cor pulmonale: echocardiogram, EKG, brain natriuretic peptide (BNP) in blood.
Hirschsprung disease: barium enema and biopsy of the rectal mucosa in patients with constipation or abdominal distention
Cardiac arrhythmias: Holter monitor.
Tumor of the neural crest: chest and abdominal x-rays when there is suspicion of neural tumor.
Developmental or learning delay: complete neurocognitive evaluation.
The principal objective in the treatment of CCHS is to ensure adequate oxygenation and ventilation during sleep and wakefulness to improve the long-term diagnosis by reducing the risk of developing cor pulmonale, PAH and neurological damage secondary to chronic hypoxia1,4,5.
Management of neural tube development defects and autonomic dysfunctions is complex and requires the participation of multidisciplinary teams.
3.3. Ventilatory Support
Because in children with CCHS the ventilatory response to hypoxemia and hypercapnia does not improve with growth or with the use drugs that stimulate the respiratory center, use of some type of a ventilatory support system is required35. These devices should be adjusted to obtain PaCO2 levels between 30 and 40 mmHg and oxygen saturation >95%5. There are various types of ventilation devices. Positive pressure ventilation is usually used with tracheostomy and/or NIPPV mask. Ventilatory parameters should be carried out via PSG in which correction of gas exchange during sleep can be confirmed.
• Positive pressure ventilation by tracheostomy (PPVT) is frequently used in infants and small children. It tends to be used during ages until maturation of the respiratory apparatus and CNS is achieved. It is considered to be the safest management for guaranteeing adequate ventilation and oxygenation. Portable equipment with an internal battery is necessary. At least two trained relatives are required in the use of the equipment and care of the tracheostomy. Use of tracheostomy tubes without balloon is recommended to allow for a leak sufficient for the use of a Passy–Muir® valve that stimulates development of speech and prevents subglottic stenosis. Similarly, the equipment should be capable of compensating for the leak and in turn guarantee an adequate pulmonary insufflation5,36,37.
• Non-invasive positive pressure ventilation (NIPPV) is done through nasal or oronasal masks. It is less costly and easier to use than the PPVT; however, it is not advisable in patients who require 24-h ventilation or high pressures. Prolonged use of masks for NIPPV may cause skin lesions and facial hypoplasia in very young patients. Because patients with CCHS cannot adequately trigger the ventilator, the ventilatory mode used should be able to produce a back-up respiratory frequency. It is also useful for replacement of the PPVT in patients with stable clinical course and who are able to cooperate with the use of NIPPV—usually during school age, and in those in whom removal of the tracheostomy cannula is planned. In those cases, the pressures required for ventilation tend to be greater than those used in PPVT because the NIPPV should overcome the resistance of the upper airway38-41.
• Other ventilatory support systems—use of negative pressure ventilation systems using vests or chest covers42 as well as diaphragmatic pacemakers has been described. The latter tend to be associated with NIPPV, and both devices could be alternated during the day and night, respectively43-45.
The use of ventilatory support requires periodic medical follow-up and adjustment of the ventilatory parameters with PSG at least once a year, especially in young children due to their growth.46 Children with CCHS have a higher risk of presenting neurocognitive disorders that make learning difficult.47 In adolescents, use of alcohol and drugs should be monitored as these substances could further depress respiration and increase the risk of death.48,49 Inadequate ventilation management also increases the risk of damage caused by hypoventilation and chronic hypoxemia.50
The only risk factor associated with mortality is gender: males present a higher risk. In patients without ventilatory support the prognosis is poor, especially with elevated mortality rates during the first 3 months of life, which confirms that the respiratory center is more unstable at this age and that ventilatory support should be instituted as soon as there is diagnostic suspicion.51 Global mortality according to different series varies between 8 and 38%. The main causes of death are pneumonia, decompensated cor pulmonale and aspiration; mortality is higher in patients without ventilatory support or with inadequate support52,53.
CCHS is a rare genetic disease, but has shown to be more common in neuropediatric services and sleep clinics. The disease requires a high level of suspicion in patients (especially pediatric) with hypercapnic respiratory insufficiency without an apparent origin. Early diagnosis is important for the initiation of ventilatory support, which will help to prevent the development of complications as well as to reduce mortality.
Ethical disclosure
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of data. The authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consent. The authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Funding
None.
Conflict of interests
The authors declare they have no conflicts of interest.
Received 31 March 2015;
accepted 2 July 2015
* Corresponding author.
E-mail:jlcarrillo14@hotmail.com (J.L. Carrillo-Alduenda).