





The synthesis progression of the crystalline silica polymorph tridymite has been investigated through the crystallization evolution using X-ray diffraction (XRD) technique. The impact of various synthesis variables on the crystallization of tridymite was examined, including the crystallinity of the raw material, the type and concentration of the mineralizer used, as well as the effect of the maximum furnace temperature and soaking time. The evolution of the crystal structure was analyzed using X-ray diffraction, and the best-synthesized final product was characterized by X-ray fluorescence, scanning electron microscopy and specific surface area. Due to the inherently sluggish nature of the tridymite transition, it was not possible to synthesize tridymite without the presence of an alkali in the composition. While all the variables studied influenced the synthesis of tridymite to some extent, the concentration and nature of the nucleation agent were identified as the key parameters to optimize the synthesis process. As a result of this research, a synthetic silica product with high crystalline tridymite phase content was successfully obtained, which could be deemed suitable for use as a secondary reference material in the quantification of crystalline silica, given the lack of reference materials for this kind of structure.
En el presente trabajo se ha evaluado el proceso de síntesis del polimorfo de la sílice cristalina tridimita a través de la progresión de la cristalización de su estructura cristalina mediante la técnica de difracción de rayos X. Las variables de síntesis seleccionadas fueron la cristalinidad de la materia prima, el tipo y concentración del mineralizador utilizado, así como el efecto de la temperatura máxima de horno y el tiempo de permanencia durante el proceso de síntesis. La evolución de la formación de la estructura cristalina se siguió a través de su capacidad de cristalización por difracción de rayos X y el producto final optimizado con la estructura deseada se caracterizó, además, por fluorescencia de rayos X, microscopio electrónico de barrido y superficie específica. Debido a la naturaleza poco activa de la transición de la tridimita, no se obtuvo tridimita con elevada cristalinidad sin la presencia de un ion alcalino en la composición. Aunque todas las variables estudiadas ejercieron cierta influencia en la síntesis de la tridimita, la concentración y naturaleza del mineralizador fueron los parámetros clave para la optimización del proceso de síntesis. Dicha investigación permitió obtener un producto sintético de tridimita de elevada calidad y grado de cristalinidad que podría considerarse como candidato para ser utilizado como patrón secundario en la cuantificación de sílice cristalina, dado la falta de materiales de referencia para este tipo de estructura.
In the past year, significant developments regarding crystalline silica management in ceramic products have occurred. Regulatory agencies have intensified their efforts to enforce stricter exposure limits due to growing concerns about its health impacts, particularly respiratory diseases like silicosis and lung cancer when final product is manipulated [1–3]. Respirable silica is released into the air when using any of the dusty materials or when machining solid materials containing crystalline silica [4].
Ceramic products, minerals, ores, gravel, sand and other natural products containing crystalline silica are exempt from the REACH regulation (Registration, Evaluation, Authorization, and Restriction of Chemicals, 1907/2006), provided they are not chemically modified [5]. Despite this, Industrial Mineral Producers (IMA), have collectively decided to classify even these non-modified products with crystalline silica based on their crystalline silica content, following a Hazard Assessment [6]. As a result, products with a respirable crystalline silica concentration of 10wt% or more should be labelled as STOT RE 1 (Specific Target Organ Toxicity upon Repeated Exposure, Category 1, H372). If the concentration is between 1.0 and 10wt%, the labelling should be STOT RE 2 (Specific Target Organ Toxicity upon Repeated Exposure, Category 2, H373). No classification is required for mixtures and substances with a respirable crystalline silica content below 1.0wt%, according to IMA guidelines.
These developments underscore the critical importance of maintaining a constant vigilance and innovation in managing crystalline silica in the ceramic industry. Because of these events, the quantification of crystalline silica in ceramic tiles is presented as an important analytical challenge, with the aim of establishing safer and healthier working environments [7,8].
Crystalline silica includes the silica minerals quartz and its polymorphs such as cristobalite and tridymite, which have the same chemical formula but different crystal structures. Amorphous, non-crystalline silica can also occur in a wide variety of geologic environments [9,10]. The crystal structure of all these silica polymorphs is made up of a three-dimensional framework of SiO4 tetrahedra, which are linked together by sharing the corners with other tetrahedra. In these lattices, therefore, each oxygen atom has two silicon atoms as nearest neighbours, and each silicon atom is surrounded by four oxygen atoms. Despite these similarities, each silica polymorph has its own crystal structure [11]: in tridymite the silica tetrahedra are packed in a two-layer structure meanwhile in the cristobalite they are organized in a three-layer structure [12]. The more open crystalline structure of cristobalite and tridymite in comparison to quartz allows a certain degree of incorporation of other elements into the crystal structure.
The three crystalline silica polymorphs can occur inherently in ceramic tiles due to their capacity to crystallize during the high thermal cycles they are subjected to in the production process (above 1100°C). Generally, the cristobalite and tridymite developed naturally together and they are formed by heating silica to high temperatures [13–15]. Obtaining isolated tridymite is very strange and it may actually not exist as pure SiO2. All natural tridymite contains small amounts of alkali, being practically impossible to synthesize without their presence using a mineralizer due to the drowsy nature of its transition [13].
Mineralizers are defined as inorganic substances that accelerate the rate of a process or reaction occurring in the solid state, within the liquid phase, or at the solid-liquid interface. The term mineralizer primarily applies to minor components that stimulate the formation of the principal silicate, which in this case is crystalline silica. A flux is understood to be a substance capable of reducing the temperature at which the liquid phase forms. Many substances that act as mineralizers are also high-quality fluxes for the raw materials involved in the synthesis process. Some of the selected mineralizers used in the present study have already been used in previous research carried out by Alharbi (LiF) [13], Kather (CaF2) [16], Moseman (NaCO3) [17], Lambotte (NaF) [18] and Dapiaggi (NaOH) [19].
Although there are many studies dealing with the transformation of amorphous silica to tridymite and cristobalite at around 1000°C [20,21], quite a few studies have examined the quantities and characteristics of these silica crystalline phases when they are processed at high temperatures [19,22].
Existing standard on the quantification of crystalline silica as NIOSH 7500 [23–25] talk about certain reference material regarding determination of tridymite in bulk samples. But the product mentioned is out of stock all around the world and there is no signal of another reference materials available to carry out the quantification test.
At present, there is no harmonised procedure for the determination of free crystalline silica in ceramic tiles [26]. The variation of silica crystallinity and possible formation of its polymorphs during the high temperature synthesis process [27], the presence of interferences between crystalline structures [28] and the non-availability of certified reference materials (CRM) for all silica polymorphs (tridymite) are the main causes. The need for standards is a common element in any analytical technique to endorse the correct performance of the test and to guarantee the comparability of the results obtained between centres.
In this context, the present work focuses on the development of a synthesis process to develop tridymite, with the aim of obtaining a high-quality product that meets the necessary crystallinity characteristics to be able to establish by itself as a reference material to quantify effectively the whole crystalline silica content in ceramic products.
During the development of the methodology, different tridymite synthesis variables have been studied, such as the influence of the raw material used, the type and concentration of mineralizer selected as well as the maximum temperature and the soaking time of the synthesis process.
Regarding the methodology selected, to quantify the crystalline silica content in ceramic tiles and ensure compliance with regulatory standards and product specifications the most commonly used method is X-ray diffraction (XRD), which is able to distinguish and quantify the different silica polymorph present in a sample. This method has been selected to follow the crystallization progress of the tridymite structure all along the present study. Another testing method is Fourier transform infrared spectroscopy (FTIR), which analyses the infrared absorption spectrum of a sample [29–32]. Nevertheless, the technique is limited in their capacity to distinguish between silica polymorphs, especially in the presence of silicates.
Materials and methodsIn this section the description of the raw materials selected, the preparation of compositions and the test methodologies used to carry out tridymite synthesis are detailed. Additionally, all characterization techniques used to carry out the present study are described.
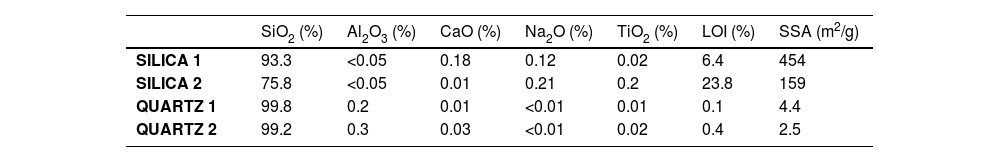
MaterialsTo develop the research four different commercial raw silica materials were selected whose main common characteristic was presenting high-purity silica. The main difference among them was their crystalline structure. On one hand, two products were amorphous silica referenced as silica 1 (Silica Supelco©) and silica 2 (Silica Siloid©). On the other hand, the other two selected raw materials were fine-grain quartzes (d50<5μm) referenced as quartz 1 (EL01) and quartz 2 (Min-u-sil5©).
Selection of raw materials was based on previous studies made by Alharbi [13], Dapiaggi [19] and Venezia [20] which stated the influence of crystallinity and purity in the synthesis of high-temperature silica polymorphs.
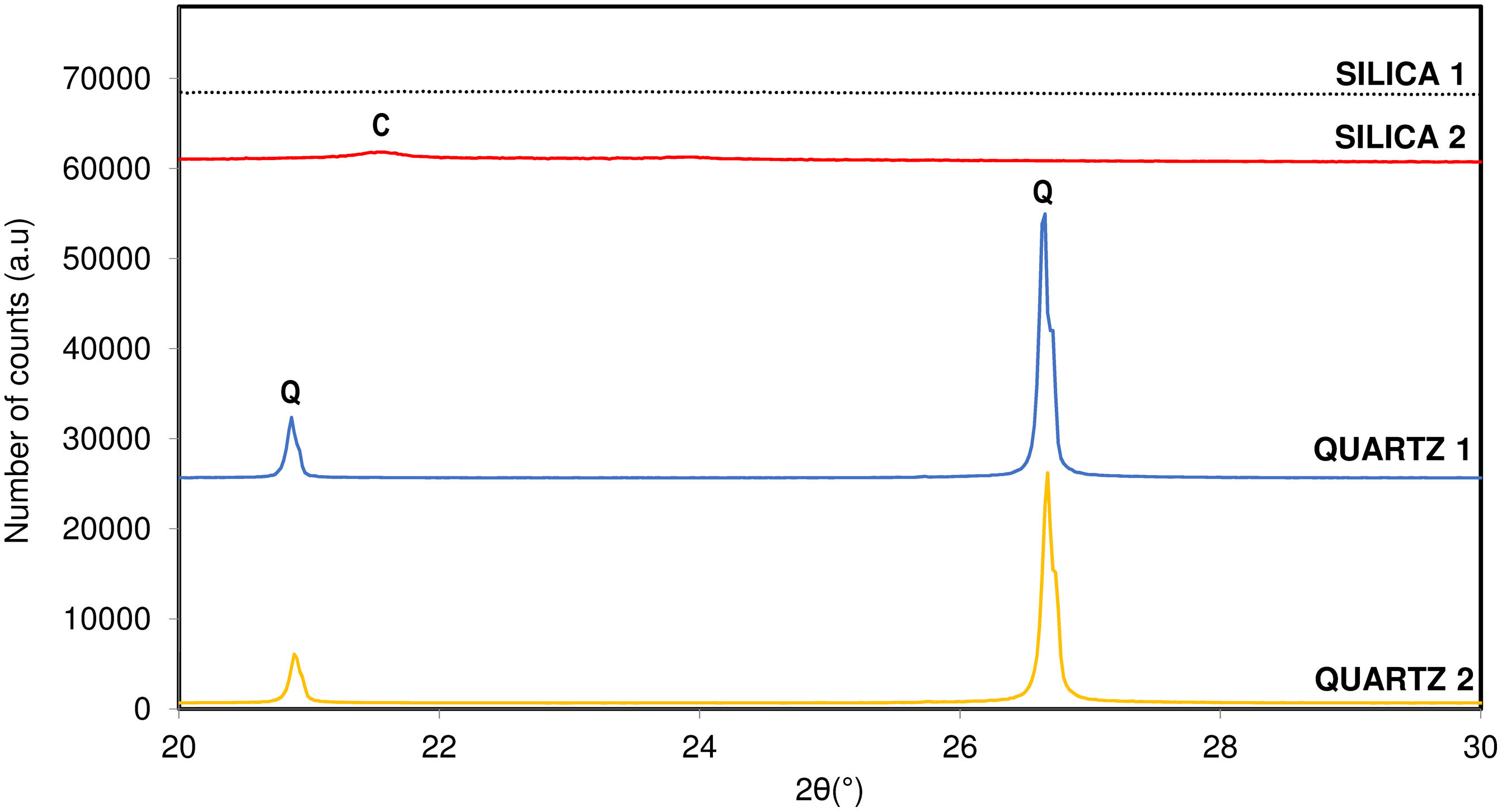
In Table 1 physico-chemical characterization carried out on each raw material are shown. Fig. 1 shows the different crystallinity of the selected materials characterized by X-ray diffraction.
Physico-chemical characterization of raw silica materials (%).
| SiO2 (%) | Al2O3 (%) | CaO (%) | Na2O (%) | TiO2 (%) | LOI (%) | SSA (m2/g) | |
|---|---|---|---|---|---|---|---|
| SILICA 1 | 93.3 | <0.05 | 0.18 | 0.12 | 0.02 | 6.4 | 454 |
| SILICA 2 | 75.8 | <0.05 | 0.01 | 0.21 | 0.2 | 23.8 | 159 |
| QUARTZ 1 | 99.8 | 0.2 | 0.01 | <0.01 | 0.01 | 0.1 | 4.4 |
| QUARTZ 2 | 99.2 | 0.3 | 0.03 | <0.01 | 0.02 | 0.4 | 2.5 |

The effect of different high-grade purity nucleant agents from Sigma Aldrich© was studied. The selection was made according to literature information which points out that crystallization process can be influenced either by the cation and the anion nature. Several studies have been published showing the influence of using mineralizing agents either alkali or alkaline earth nature to obtain tridymite [13,16–20].
Experimental methodThe experimental procedure used in the laboratory to study the synthesis capacity of tridymite begins with dosing the synthesis components: the silica raw material and the mineralizer. The concentrations of the mineralizers used in the present research ranged from 2% to 12%.
The next step involved grinding and mixing in a McCrone mill to ensure intimate contact between the components. Agate pieces were used as grinding elements to prevent contamination and avoid introducing new cations to the mixture. The grinding process was conducted in a 30-min cycle at the maximum speed of the equipment.
The ground mixture was then placed in an alumina crucible with a lid and sintered under a thermal cycle determined by the study stage. Sintering was performed in a Pyrometrol electric furnace (HP5), with maximum temperatures ranging from 850°C to 1400°C and soaking time from 2 to 32h to study the effect of the heat treatment.
After the synthesis process, the final product was disaggregated in an agate mortar until a particle size of less than 100μm was achieved, facilitating the study of crystallinity based on XRD crystallite theory.
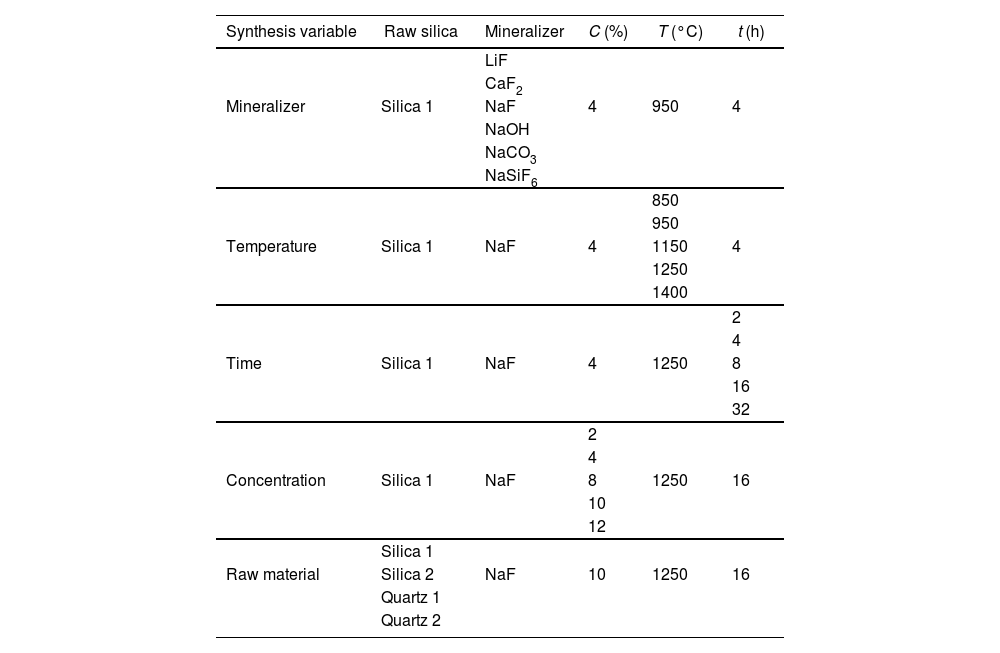
In Table 2 a summary of synthesis conditions selected is shown to comprehend the study developed. Process parameters that can present certain influence on the tridymite synthesis were selected attending previous research carried out by the authors [26].
Summarize test parameters selected to study influence of synthesis variables.
| Synthesis variable | Raw silica | Mineralizer | C (%) | T (°C) | t (h) |
|---|---|---|---|---|---|
| LiF | |||||
| CaF2 | |||||
| Mineralizer | Silica 1 | NaF | 4 | 950 | 4 |
| NaOH | |||||
| NaCO3 | |||||
| NaSiF6 | |||||
| 850 | |||||
| 950 | |||||
| Temperature | Silica 1 | NaF | 4 | 1150 | 4 |
| 1250 | |||||
| 1400 | |||||
| 2 | |||||
| 4 | |||||
| Time | Silica 1 | NaF | 4 | 1250 | 8 |
| 16 | |||||
| 32 | |||||
| 2 | |||||
| 4 | |||||
| Concentration | Silica 1 | NaF | 8 | 1250 | 16 |
| 10 | |||||
| 12 | |||||
| Silica 1 | |||||
| Raw material | Silica 2 | NaF | 10 | 1250 | 16 |
| Quartz 1 | |||||
| Quartz 2 |
The synthesis products obtained were microstructurally characterized by X-ray diffraction (XRD), X-ray fluorescence (WD-FRX), scanning electron microscopy (SEM) and nitrogen gas adsorption (NGA).
Chemical composition was conducted with a PANalytical model AXIOS wavelength dispersive X-ray fluorescence (WD-XRF) spectrometer with a Rh anode tube, and 4kW power, fitted with flow, scintillation, and sealed de-tectors, eight analysing crystals: LiF200, LIF220, Ge, TLAP, InSb, PE, PX1 and PX7, and provided with masks of 37, 30, 27, 10, and 6mm in diameter.
Identification of the crystalline phases was monitored by an X-ray diffractometer (XRD) Theta-Theta D8 Advance A25 from Bruker with CuKα radiation (λ=1.54183Å) [26–29]. The generator settings were 45kV and 30mA. The XRD data were collected in a 2θ of 5–90° with a step width of 0.015° and a counting time of 1.2s/step by means of a LinxeyEYE detector. The quantification of crystalline phases was using the collected data were determined in a Rietveld refinement. The 6.0 version of the Rietveld analysis programme DIFFRACplus TOPAS was used, assuming a pseudoVoight function to describe peak shapes. The refinement protocol included the background, the scale factors and the global-instrument, lattice, profile and texture parameters. The basic approach is identifying all the crystalline phases present and input basic structural data for all phases, and then let the computer model the data until the best fit to the experiment pattern is obtained. An internal standard with a known concentration was introduced in every powder analyzed sample. Rwp (R-weighted pattern) and GOF (Goodness of fit) parameters were calculated in order to evaluate the accuracy of the diffractogram simulation. Diffraction patterns used to identify and quantify crystalline phases present all along the study belong to the International Centre of Diffraction Data (ICDD©) database 2024: Tridymite: ICDD PDF4+04-025-7001 for Tridymite, ICDD PDF4+04-006-2056 and ICDD PDF4+04-003-6495 for Quartz.
The determination of microstructure was carried out using an FEI model FEG-ESEM Quanta 200 field-emission environmental scanning electron microscope (SEM) equipped with an energy dispersive X-ray microanalysis [Si(Li) EDAX Genesis 7000 SUTW (super ultrathin window)]. Analyses were performed at 20kV (beam voltage) in the high vacuum mode (∼10−6mbar). Acquisition time of the spectrums was 300s, and the detector dead time was of about 30%.
Micrographs of the samples were taken with the backscattered and secondary electron signals from a field emission scanning electron microscope under high vacuum conditions (high resolution). The backscattered electron signal provides information on topography and composition. It is more intense the higher the average atomic number of the sample, so that lighter areas contain heavier elements (compositional contrast). The secondary electron signal is shallower so that it provides information on the morphology of the sample, highlighting surface irregularities such as cracks, pores, grain or crystal boundaries. Both signals provide complementary information, allowing each individual particle to be properly defined, thus reducing the error that could result from misattributing the boundary between them.
The samples were also analyzed with an energy dispersive X-ray microanalysis (EDX) equipment connected to the microscope. The interaction volume of the electron beam is in the order of 3μm or more, so that chemical information from the surroundings can be received when analysing very small areas. In addition, it should be noted that elements of atomic number equal to or greater than 6 (from carbon onwards) are detected with this analysis system. The samples were deposited on aluminium sample holders coated with conductive carbon adhesive. The samples were then coated with platinum.
The specific surface area (SSA) was determined, using nitrogen gas as adsorbent, by means of an adsorption/desorption equipment TriStar 3000 from Micromeritics, applying the standard ISO 9277:1995. Specific surface area parameter was determined applying the multi-point BET method on the adsorption curve. The amount of nitrogen adsorbed was measured by means of a static volumetric method. Before carrying out the test, the sample was dried in an oven at 60°C for 2h and, after that, it was out gassed with a nitrogen continuous flow at 60°C.
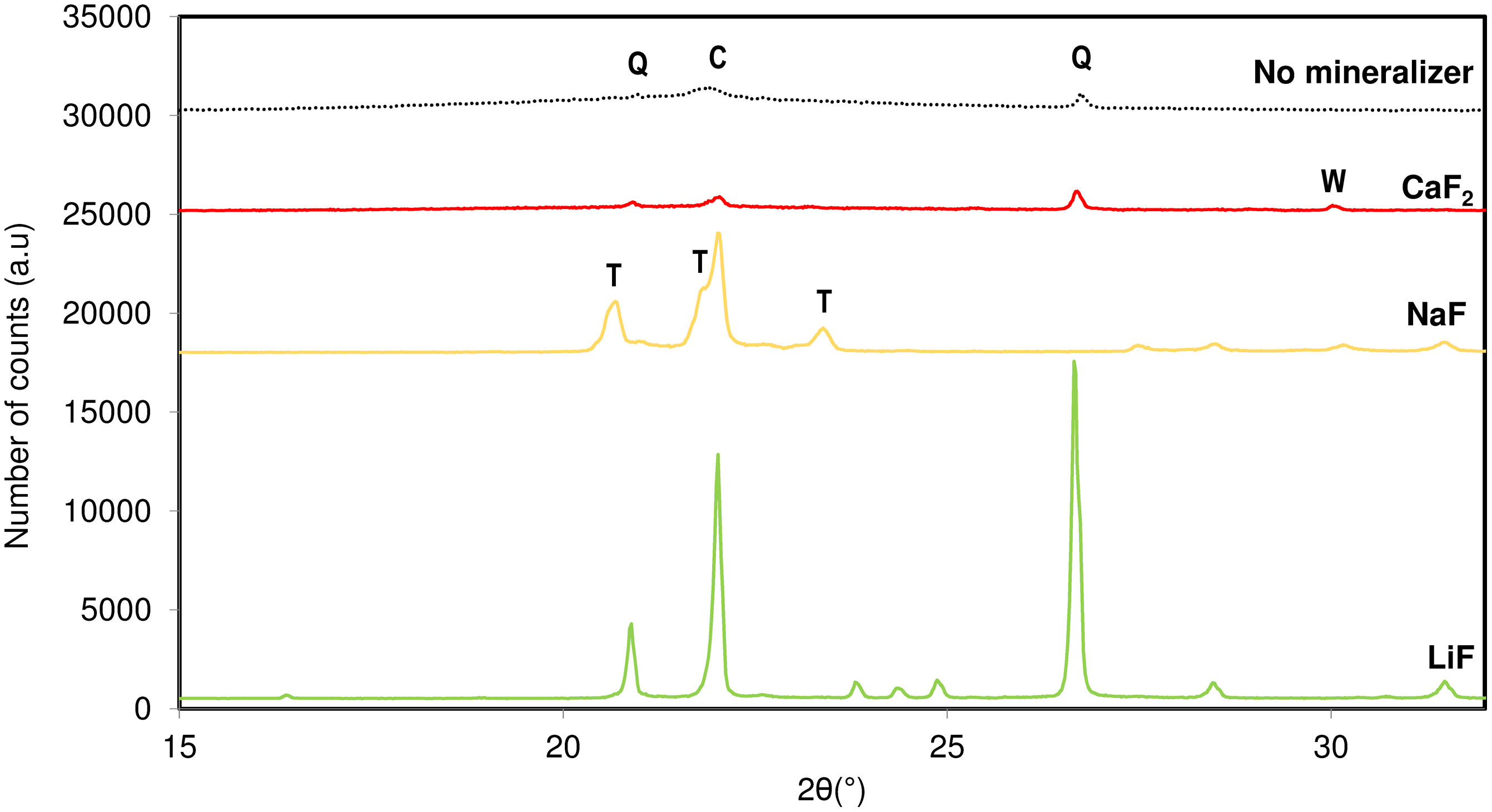
Results and discussionInfluence of mineralizer natureIn Fig. 2, the crystalline phases formed after the addition of different fluorinated mineralizers to an original amorphous silica composition (Silica 1) at a synthesis temperature of 950°C are shown. The experiments were conducted while keeping the rest of the synthesis variables constant.
. C: Cristobalite, Q: Quartz, T: Tridymite, W: Wollastonite.")
In this study, mineralizers from the fluoride group, such as lithium, sodium and calcium fluoride, were selected to act as nucleant agents. The presence of fluorinated salts accelerated the formation of significant crystalline silica phases due to a reduction in the lower stability limit of the phase in the mixture [16,18]. This effectiveness is mainly due to the presence of fluoride anions, which have high electronegativity and interact with the Si4+ cations in the solid phase, increasing their ionic mobility. This surface remains in a state of maximum energy, allowing for the polarization and reorganization of atoms, which increases the solid activity, both for forming new phases and for melting the material.
The influence of fluoride-containing salts during the firing stage varies considerably with the nature of the cation combined with the fluorine. The effectiveness of cristallizability of NaF when combined with raw materials below 1280°C is generally greater compared to other mineralizers like LiF. This could be explained to the fluoride effect, which improves tridymite crystallinity significantly as the atomic number of the combined cation increases when the cation presents an alkaline nature. This results in a higher formation of tridymite, which is more favourable at these temperatures. The presence of sodium as cation in the composition helped in breaking up the network structure at low energy and led to form new covalent bonds in the amorphous flux [13].
Nevertheless, it would not justify the effect observed for CaF2. In the case of the alkaline earth fluoride, the volume of the cation is so massive that it cannot be inserted in the tridymite structure. On the other hand, this nucleating agent showed the trend to crystallize towards another crystallization system, observing the capacity to nucleate wollastonite as a mineral phase. The fact of being facilitated the melting process decreasing the viscosity of the liquid flux formed, in conjunction with the presence of Ca2+ ions, led to crystalline structures non-desired for the present study.
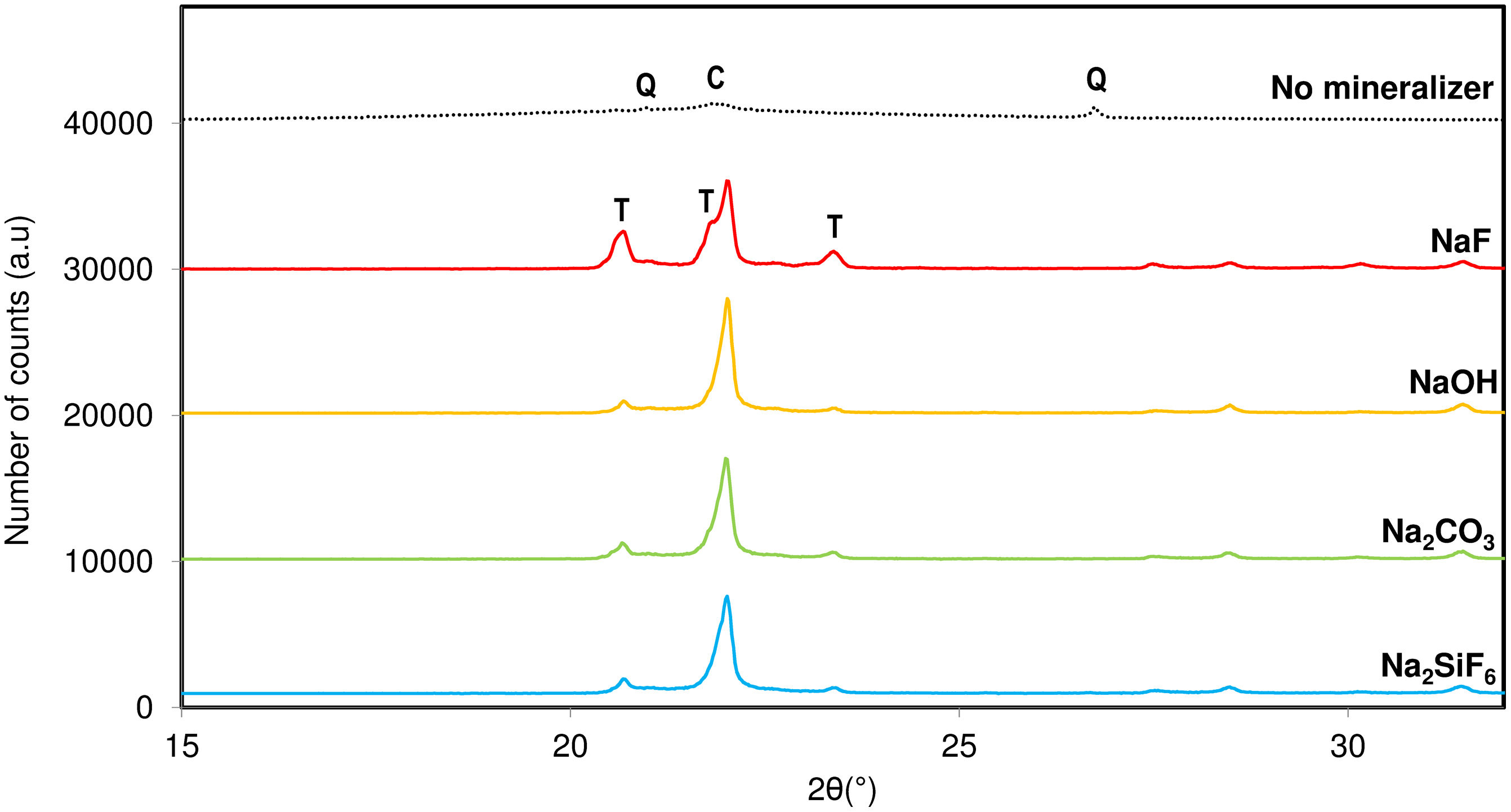
Fig. 3 shows the devitrification of silica crystalline phases using different nature mineralizers under the same alkaline cation. Sodium fluoride, hydroxide, carbonate and fluorosilicate were used in the synthesis process maintaining constant the rest of the variables. At the light of the results, it was observed that F− is the anion that better favoured the crystallization of tridymite. As mentioned before, fluorinated salts can be an important melting ageing which increases the driving force for the formation of tridymite since it can favour a reduction in its activation energy. The rest of the mineralizers presented certain trend to crystallize tridymite, but by far much lower than the fluorine.
. C: Cristobalite, Q: Quartz, T: Tridymite.")
The results have shown that addition of fluoride mineralizers result in a marked lowering of the temperature at which crystallization can begin, widening of the crystallization range because there is a reduction of viscosity of the melting phase. This phenomenon can be attributed to the weakening of the network structure brought about by fluorides. Other theories stated by Khater [16] indicates that fluorides are known to be immiscible in silicate melts, leading to two-phase glass separation generating numerous droplets that reduces the energy barriers necessary for crystallization.
Consequently, NaF was selected as the effective mineralizer for the rest of the study because it exhibited a high fluxing and mineralizing capacity in the mixture, promoting the devitrification of tridymite in the synthesis at 950°C [18]. NaF seemed to favour the thermodynamic stability of tridymite and cristobalite compared to other fluorides, which deviate the system towards the devitrification of quartz or cristobalite. NaF allowed for the formation of tridymite at temperatures below 1000°C, effectively reducing the liquid phase formation temperature of silica by approximately 200°C, thereby decreasing viscosity and surface tension.
Figs. 2 and 3 present the minimum synthesis conditions under which the presence of tridymite in the final product was observed. This presence allowed for the selection of the nucleant agent to be used all along the present research.
Fig. 4 shows the microstructures of the sample with (Fig. 4b) and without the addition of NaF (Fig. 4a) in its composition during the synthesis process. In the original sample, no degree of crystallization is observed. In micrograph b, at the same synthesis temperature, some crystallinity is observed, although it is not possible to distinguish the presence of tridymite from the formation of cristobalite also present in the final product. The micrograph with lithium fluoride is not presented, as it promoted the devitrification of quartz, which was not the focus of this study.
Influence of synthesis temperature Silica 1 sintered at 950°C, (b) Silica 1 sintered at 950°C with 4% NaF as mineralizer.")
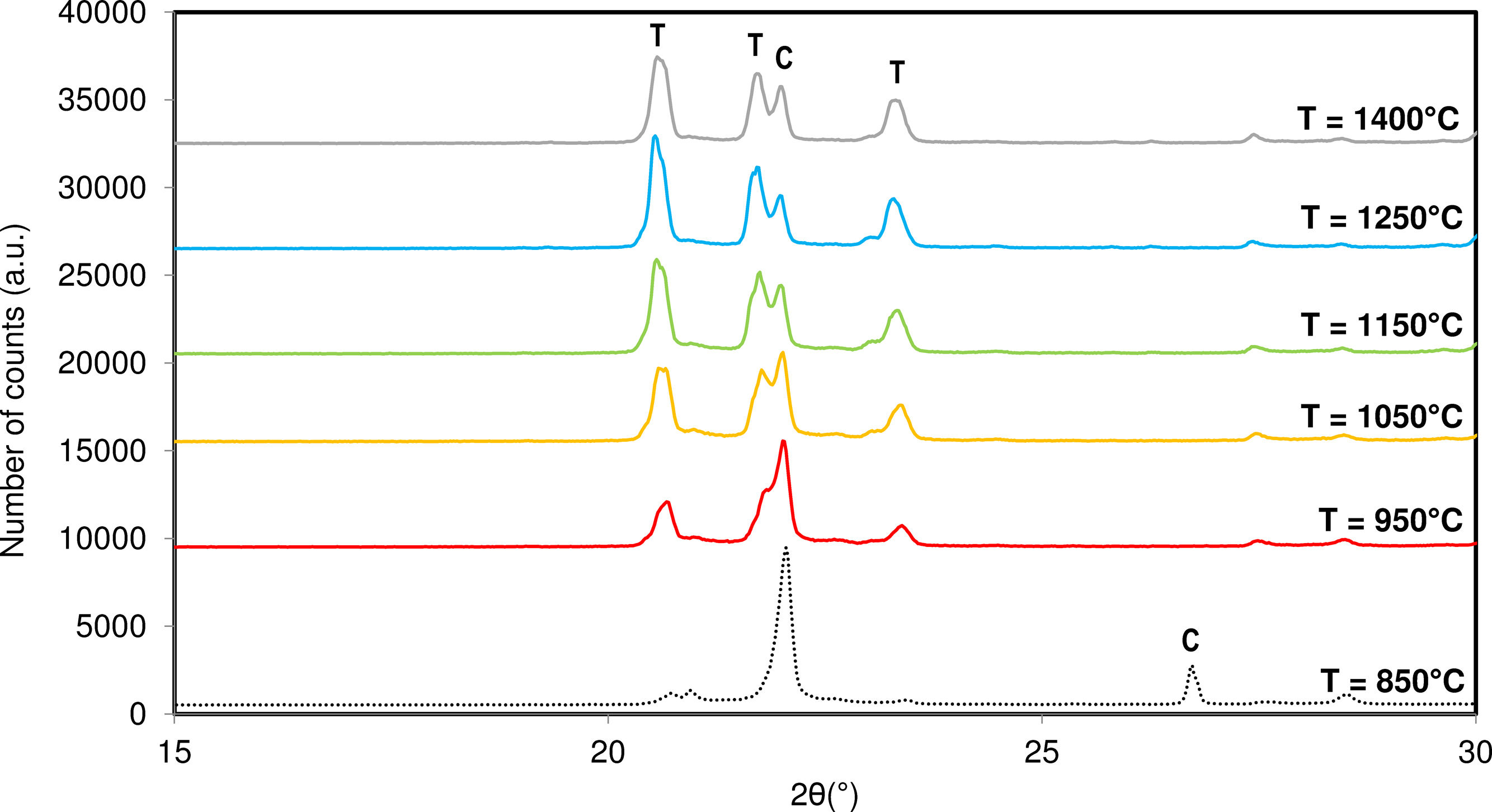
Fig. 5 shows the progression of the crystalline structure of the system influenced by the maximum synthesis temperature. It was observed that at lower temperatures, the system tends towards the crystallization of primarily cristobalite. However, as the synthesis temperature increases, the system appears to favour the formation of tridymite. The increase in temperature reduces the viscosity of the system's liquid phase and enhances the diffusion of Si4+ ions through the crystal lattice, promoting the formation of tridymite over cristobalite by providing sufficient energy to compensate for the low stability of this structure.
. C: Cristobalite, T: Tridymite.")
Higher temperatures led to an increase in the mobility and diffusion of the different ions in the structure promoting the tridymite crystallization instead of cristobalite.
However, beyond 1250°C, no increase in tridymite formation is observed. At this temperature, the viscosity of the system no longer favours the crystallization of the tridymite phase. It seems that the system tends towards material fusion, and the diffraction peaks lose intensity and broaden, forming a more amorphous and less crystalline structure.
Although an optimal synthesis temperature is observed, it is not possible to obtain a pure tridymite structure solely by modifying the firing cycle.
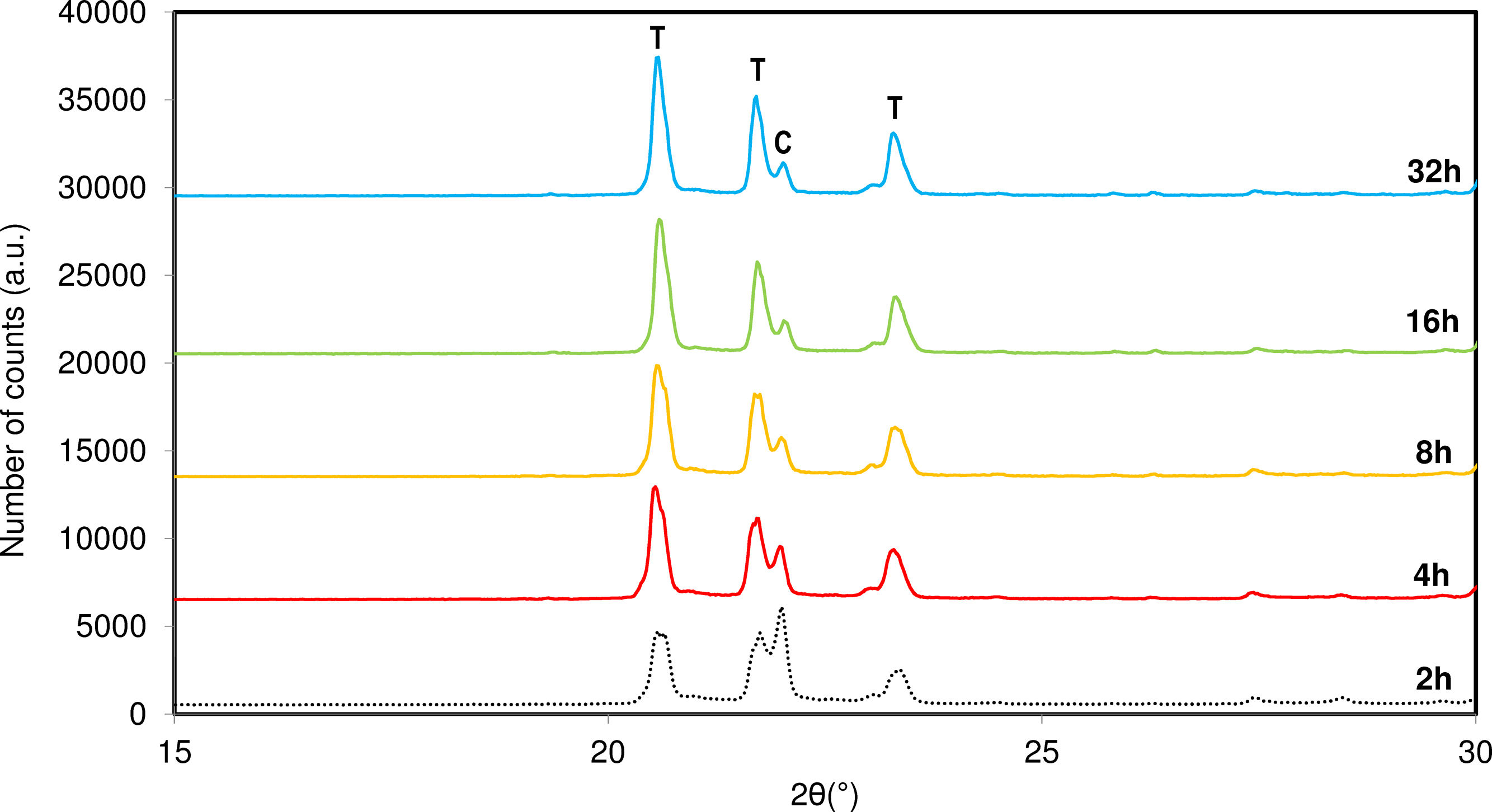
Influence of soaking timeAfter optimizing the synthesis temperature, the influence of soaking time was studied. Synthesis times ranging from 2 to 32h were investigated. Fig. 6 shows a comparison of the diffractograms obtained with varying soaking times.
. C: Cristobalite, T: Tridymite.")
Based on the results, it is observed that increasing the synthesis time at the maximum temperature favours the reorganization process of the system and increases the crystallinity of tridymite. An increase in time reduces the formation of cristobalite in the system and enhances the crystallinity of tridymite, maintaining the system at the optimal temperature for tridymite formation.
This behaviour suggests that the ions require a certain amount of reorganization, and the synthesis of tridymite does not occur instantaneously. Instead, the reaction rate in the solid state necessitates significant time for adequate ion diffusion into the developing crystalline structure. When the system is allowed to establish appropriate fluid phase viscosity conditions by adjusting temperature and time, it experiences a reduction in reaction enthalpy, leading to increased ion ordering and the development of the desired structure.
Influence of mineralizer concentrationAlthough temperature and soaking time may help system to crystallize into tridymite structure, they are only physical parameters to control synthesis progression and they do not apply enough driving force to promote pure tridymite crystallization. It can be supposed that only by using chemical actions, it would be possible to minimize the presence of cristobalite in the system.
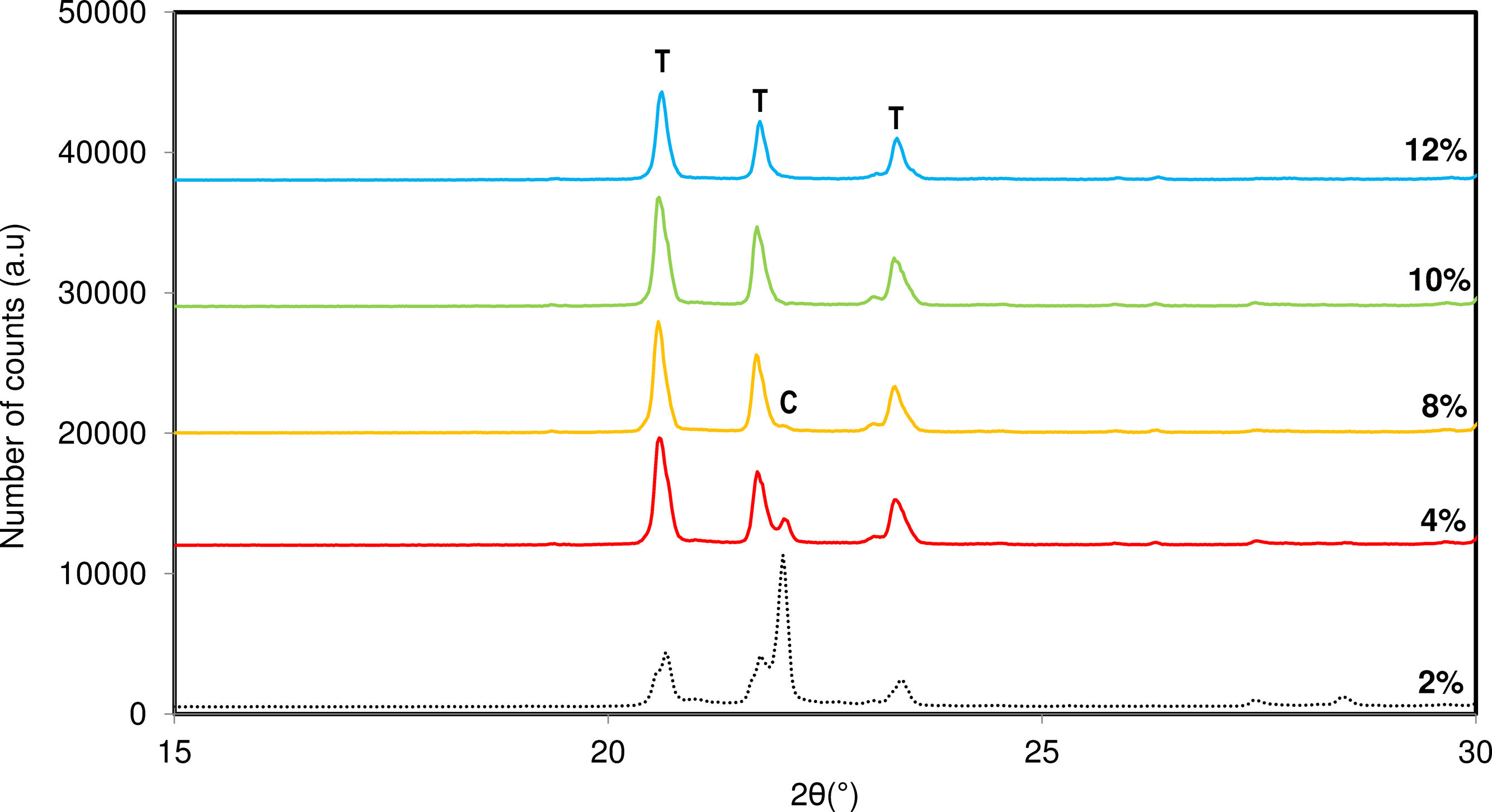
In previous sections, the influence of the mineralizer nature was studied. However, given the favourable effect of fluorides on the crystallization of tridymite, it was necessary to optimize the percentage of mineralizer required to obtain only tridymite in the final product. Fig. 7 shows the diffractograms obtained as a function of the mineralizer concentration in the original sample.
. C: Cristobalite, T: Tridymite.")
According to the results, it seems to be an optimal percentage of mineralizer that minimizes the formation of cristobalite in favour of tridymite formation. In this case, the use of 10% mineralizer in the mixture neutralizes the cristobalite synthesis capacity, evolving the entire system towards the formation of a tridymite-type structure.
However, at concentration higher than 10%, the obtained diffractogram shows less intense and broader diffraction peaks, indicating that the system experiences a loss of structural ordering and amorphization of the crystalline system, which is also undesirable for developing a specific structure.
Based on the observed effects, it means there is an optimal percentage of mineralizer in the studied mixture, determined by the inflection point between avoiding the presence of cristobalite and reducing the crystallinity of the formed tridymite. This optimal percentage was established at 10% of NaF.
Formation of tridymite may be governed by the percentage of sodium in the composition since the highest sodium the lowest energy formation the low symmetry tridymite structure.
Influence of crystalline structure of raw materialThe last variable studied regarding the crystallization capacity of the tridymite structure was the influence of the crystallinity of the original raw material, composed almost entirely of SiO2. Two of the raw materials studied had an amorphous crystalline structure (SILICA), while the other two had a quartz-like crystalline structure (QUARTZ). All of them were physico-chemically characterized to determine the material's purity and specific surface area, as discussed in previous sections.
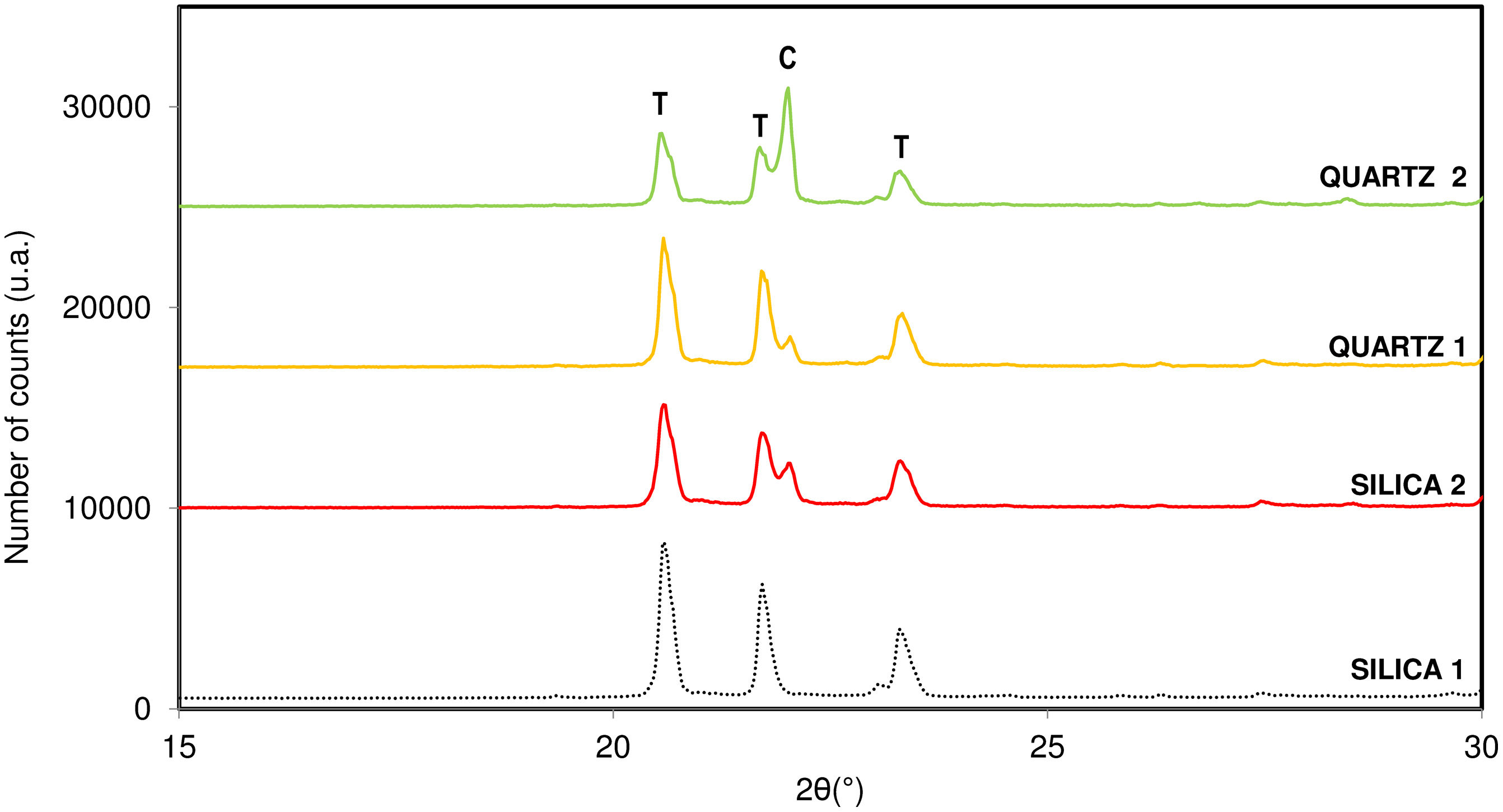
Fig. 8 presents the diffractograms obtained after carrying out the synthesis under the optimal conditions for tridymite crystallization for the rest of the variables studied.
. C: Cristobalite, T: Tridymite.")
According to the results obtained, it is observed that the sample with the greatest crystallization capacity of the tridymite structure is the one with the raw material SILICA 1. This raw material has an amorphous structure of SiO2 ions (Fig. 1) and the highest specific surface area in the entire system studied (454m2/g). On the contrary, the sample that shows the least tendency to crystallize, under the same test conditions, is the raw material QUARTZ 2, which is characterized by having the highest crystallinity of all (Fig. 1) and the lowest specific surface area (2.5m2/g), that is, a more compact and closed structure.
The observed behaviour could be related to the fact that tridymite crystallization is favoured when starting from raw materials with a more disordered and open structure and a larger specific surface area. This would indicate that the structure is weaker, and the reactivity between the raw material and the mineralizer is favoured, given the tridymite sluggish crystallization nature versus the cristobalite one.
Characterization of optimized synthesized productAfter optimizing the various synthesis variables using a raw material of ultrapure silica with low crystallinity and a sodium fluorinated mineralizer, a synthetic crystalline tridymite product was developed. This product needs to be characterized to be considered for its use as a reference material in certain crystalline silica quantifications if it is convenient.
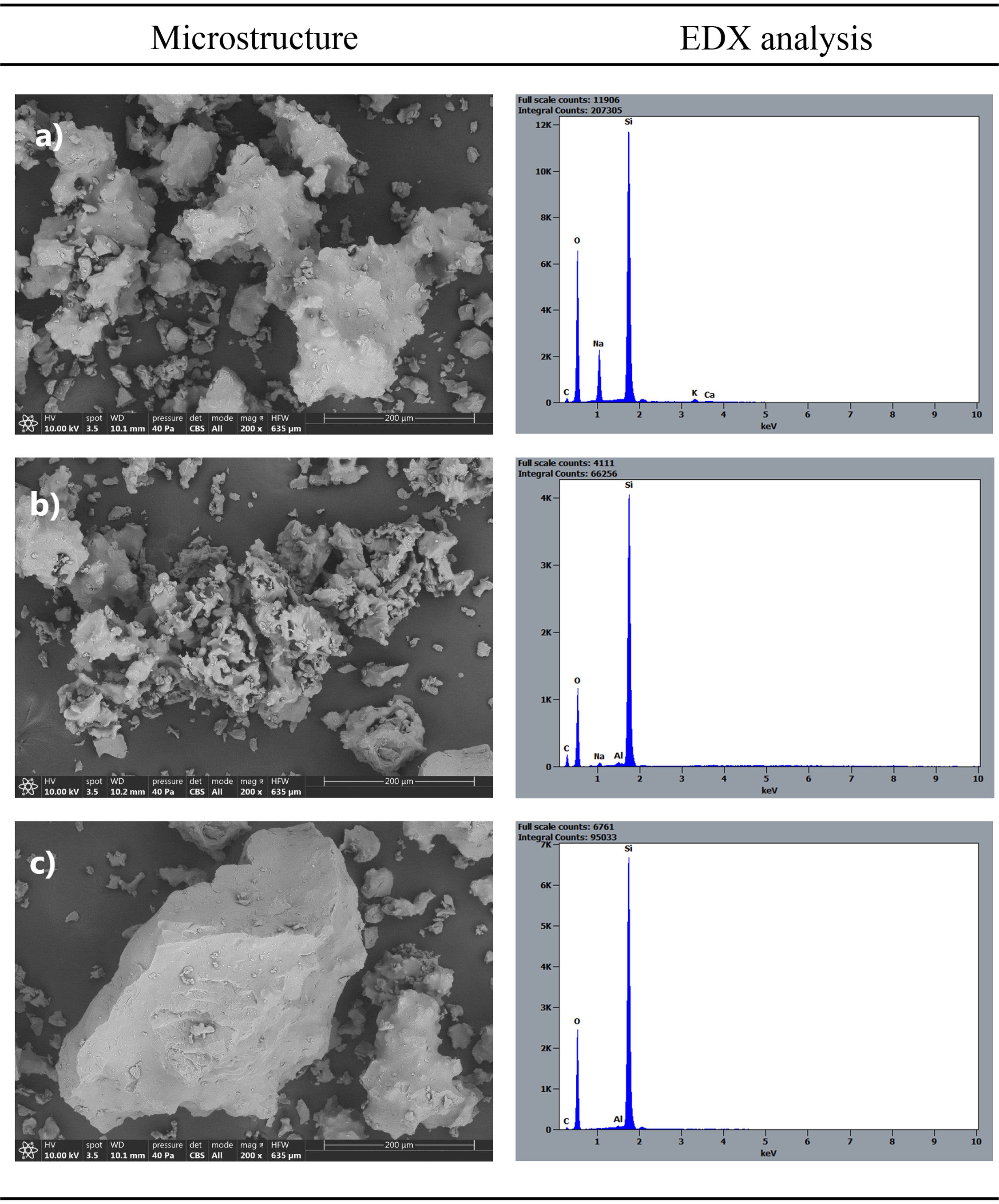
Study of microstructure of final productTherefore, the obtained synthetic tridymite product was subjected to microstructural characterization using a scanning electron microscope (Fig. 9). The micrographs revealed the presence of three types of particles or formations within the developed synthetic product. On one hand, a structure with a less crystalline appearance and a high sodium content was observed (Fig. 9a). This formation was likely related to the presence of amorphous or low-crystallinity material, representing the fluid phase that reacted but did not crystallize as tridymite. On the other hand, a type of particle with a structure of hexagonal and acicular crystals was observed, which corresponds to the tridymite signal in XRD (Fig. 9b). This kind of particles were predominant in the system, indicating the crystalline ordering of ions in a characteristic structure. The sodium content in this structure is much lower than in the amorphous material, suggesting that sodium facilitates ion diffusion through the fluid phase as a potent flux but does not integrate into the tridymite structure during crystallization. Finally, particles with edges like those observed in the original raw material were found, suggesting they might be unreacted particles from the system (Fig. 9c).
Chemical and mineralogical study low crystallinity/amorphous particles of tridymite, (b) high crystallinity-grade tridymite, (c) unreacted particles of raw silica used.")
Finally, the synthetic product was wholly characterized from a physico-chemical point of view. Firstly, the product was analyzed by XRF to determine its final chemical analysis (Table 3). Furthermore, the crystalline structures developed were quantified using the Rietveld method using an internal standard (Alumina NIST©SRM1796) to establish the final crystallinity degree of the product and determine the crystallite size developed during the synthesis process.
Physico-chemical characterization of final synthetic product.
| Chemical analysis (%) | Mineralogical analysis (%) | ||
|---|---|---|---|
| SiO2 | 94.0±0.2 | Tridymite | 98±2 |
| Al2O3 | 0.27±0.05 | Cristobalite | <1 |
| CaO | 0.21±0.01 | Quartz | <0.5 |
| Na2O | 5.0±0.1 | Crystallinity parameters (%) | |
| LOI | 0.27±0.01 | Crystallinity | 86±3 |
| F | <50mgkg−1±50 | Crystallite size | 150±25nm |
In Table 3, the results obtained after the final characterization of the synthetic product, which exhibited the best tridymite crystallinity compared to cristobalite or quartz, are presented.
According to Table 3, a tridymite synthetic product with a high grade of chemical purity was successfully obtained under the optimized synthesis conditions throughout the study. Apart from silicon oxide, it only contains a low percentage of Na2O (5%) in the composition, considering negligible the rest of the oxide contents. According to mineralogical information, the system exhibits tridymite as the only crystalline phase determined in the product, being minimized the presence of other silica polymorphs such as cristobalite or quartz. As for its structure, a high degree of crystallinity exceeding 85% has been achieved, reaching a significant challenge due to the loose nature of the tridymite polymorph and its inherently trend to be accompanied by other silica polymorphs.
The final synthetic product obtained could be considered a good candidate for selection as a secondary reference standard in the quantification of crystalline silica. Nevertheless, it must be considered that to be labelled it as a certified reference standard it would require further optimization of the crystallinity grade of the tridymite structure till reaching values higher than 90%.
ConclusionsThe objective of this work was to develop a high-quality crystalline silica product with a tridymite-type structure. To achieve this, an experimental design was created to study key variables affecting the synthesis process. These included the type and structure of the silica raw material, the mineralizer's nature and concentration, maximum temperature, and soaking time.
Results showed that using sodium cations as the mineralizer favoured tridymite formation, as the cation's size and charge improved diffusivity at temperatures below 1000°C, reducing temperature of tridymite crystallization by 200°C. Fluoride anions proved more effective in promoting crystallization than other anionic groups. Additionally, raw materials with a higher surface area and lower crystallinity enhanced tridymite formation, though this requires further study. Optimal synthesis conditions were identified as 1250°C and a 16-h soaking time, marked as inflection points in the XRD crystallization curves.
Chemical factors like the mineralizer's nature and concentration had the greatest impact on crystallization, while physical factors had a more subtle influence. The final synthetic product demonstrated no presence of other silica polymorphs in the composition as well as high crystallinity, making it a potential candidate for secondary standards in crystalline silica quantification, though further research is needed to develop a certified reference material.
Declaration of generative AI and AI-assisted technologies in the writing processDuring the preparation of this work the author(s) used [CHAT GPT] in order to improve English language and readability. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
This study has been co-financed by IVACE and the ERDF Funds, within the ERDF Operational Programme of the Valencian Community 2021–2027 in the call for aid to Technology Centres of the Valencian Community for R&D projects in cooperation with companies 2023–2024 (REACTMAC_IMDEEA/2023/81).

