





Hydroxyapatite is the main bones and teeth inorganic mineral component. This material has been an object of research aiming its use in biomaterials as bone replacement option for biomedical applications. In this investigation, the synthesis of hydroxyapatite was studied by the Hydrothermal (HT) and Microwave Irradiation (MW) methods. Here, parameters such as pH (9 and 11) and synthesis time were varied, and residue of chicken eggshell was used as calcium precursor. The obtained powders were characterized by X-ray Diffraction (XRD), Fourier Transform Infrared Spectroscopy (FTIR), Dispersive Energy X-ray Spectroscopy (EDS), and Transmission Electron Microscopy (TEM). The results showed that the material synthesized when the pH reached the value of 9 which generated less carbonated powders than those with pH of 11. The Hydrothermal method presented more satisfactory morphological results according to the TEM analysis. This procedure was considered more suitable as a route to obtain hydroxyapatite as biomaterial.
La hidroxiapatita es el principal componente mineral inorgánico de huesos y dientes. Este material ha sido objeto de investigación dirigida a su uso en biomateriales como sustituto óseo en aplicaciones biomédicas. En este trabajo, se estudió la síntesis de hidroxiapatita utilizando los métodos hidrotermal (HT) y de irradiación de microondas (MW), variando parámetros como el pH (9 y 11) y el tiempo de síntesis, utilizando como precursor de calcio, el residuo de la cáscara de huevo de gallina. Los sólidos (polvo) obtenidos se caracterizaron por difracción de rayos X (DRX), espectroscopía infrarroja por transformada de Fourier (FTIR), espectroscopía de energía dispersiva (EDS) y microscopía electrónica de transmisión (MET). Los resultados mostraron que el material sintetizado a pH 9 generó polvos menos carbonatados que aquellos obtenidos a pH 11. El método hidrotérmico presentó resultados morfológicos más satisfactorios según el análisis de MET. Este procedimiento se consideró más adecuado como una ruta para obtener hidroxiapatita como biomaterial.
Hydroxyapatite (Ca10(PO4)6(OH)2 – P63/m – Ca/P=1.67) is a calcium phosphate that has been studied for decades due to its diversity. Among some applications, it is worth mentioning biomaterials in bone grafts and dental coatings [1,2], removal of contaminants in soil and water [3], catalysis of organic compounds and others [4]. Several synthesis methods such as solid-state reaction, mechanochemical synthesis [5,6], combustion in solution, pyrolysis [7,8], sol–gel, microemulsion, hydrothermal method and microwave irradiation [9–11] have being used as techniques to obtain this phosphate.
In Hydrothermal synthesis, the reaction is conducted in the presence of water at temperature range 80–400°C, pressure up to 100 KPa and prolonged period of times [12]. The hydroxyapatite nanoparticles powders produced in this method are relatively stoichiometric and present high crystallinity. In addition, the phase purity and Ca/P ratio are significantly improved as the hydrothermal time and temperature are increased [13]. In contrast, the synthesis with microwave irradiation is characterized by the in situ microwave energy conversion into heat. Few minutes are required to reach the entire solution volume treatment temperature without any gradient [14]. The hydroxyapatite prepared by this technique, generally contains smaller particle size, good purity and a closer size distribution [13].
The choice of calcium as precursor raw material is extremely important in order to obtain the hydroxyapatite as it often influences the stoichiometry as well as the purity of the material. Chicken eggshells are mainly composed of calcium carbonate (95–97%) which can therefore be adapted as a biogenic source of calcium for the synthesis of hydroxyapatite. In addition, as it is an abundant industrial waste, its use as a raw material for calcium phosphates is a good alternative [15–17]. Other studies showed that hydroxyapatite based on this biogenic source, presents rapid biomineralization when in contact with human tissue, improved mechanical properties and superior sinterability when compared to this compound from inorganic sources [18]. Moreover, Krishna et al. [19] reported that its biocompatibility when extracted from the eggshell, favors the osteoblast cells adhesion and it is non-cytotoxic due to the CaCO3 biological nature.
When synthesizing hydroxyapatite, parameters such as pH and synthesis time can influence the material properties. Liu et al. [20], using the Hydrothermal method, fixed the synthesis temperature at 140°C and used pHs of 6, 9 and 14. The produced material exhibited stick-like dispersed particles, and the degree of agglomeration grown up as the pH increased. When using microwave irradiation synthesis, Wang et al. [21], chose temperatures of 100, 120 and 140°C and times of 1 and 30min. The obtained hydroxyapatite compound presented crystal growth proportional to time and temperature and stick-like morphologies.
In this work, hydroxyapatite powders produced by hydrothermal and microwave irradiation hydrothermal were synthesized using chicken eggshells as a biogenic source. Parameters such as pH and synthesis time were varied during the investigations.
Materials and methodsObtaining calcium precursorChicken eggshells were collected, washed by using distilled water and later had their membranes removed. Then, the shells were boiled in a mixture of distilled water and ethanol to remove impurities and organic residues followed by drying process at 100°C for 24h. The dried shells were grounded in agate mortar to produce fine particulate powder.
For the formation of Ca(OH)2, 11g of the eggshell powder was dissolved into 209mL of hydrochloric acid (HCl, PA, Neon, Brazil) at a concentration of 0.8molL−1 under stirring at 40°C for 1h. Then, an 80mL solution of 1.09molL−1 sodium hydroxide (NaOH, Macron, Brazil) was added. Finally, the precipitate was filtered and dried at 100°C in an oven for 24h.
Synthesis of hydroxyapatiteIn order to obtain hydroxyapatite powders, pH parameters, temperature and time for conventional hydrothermal and microwave irradiation synthesis were determined. This work is based on previous investigations carried out by Qi et al. [9] who varied the pH in the Hydrothermal synthesis, Méndez-Lozano et al. [22] who applied different synthesis times by Microwave Irradiation, Nga et al. [23] who used several times in the Hydrothermal synthesis, and Rivera et al. [24] who altered the pHs in the Microwave Irradiation synthesis.
The synthesis of hydroxyapatite was performed starting from equal volume precursor solutions containing 0.0279mol of calcium hydroxide (Ca(OH)2) and 0.0167mol of phosphoric acid (H3PO4). The molar ratio was kept Ca/P=1.67 and pH was adjusted to 9 or 11 with ammonium hydroxide solution (1:1). After addition of the precursors, the resulting solution was aged at 80°C under magnetic stirring for 2h. This last process was adopted on the products obtained from the two synthesis methods, so that the powders were better compared.
For the Hydrothermal Synthesis, after the aging period, the precipitate was placed into a Teflon reactor, placed in a stainless steel autoclave and kept in the oven for 6 or 12h at 180°C.
Microwave Irradiation synthesis was performed at the end of aging time, at 80°C with heating ratio of 5°Cmin−1, and synthesis times of 1 and 36minutes. A Panasonic Smart Junior microwave operated under 800W power and 2.4GHz frequency was used.
The products obtained from both methods, were filtered and washed three times with distilled water and once with ethanol to remove impurities. Finally, the solid was oven dried at 100°C for a period of 24h.
The produced samples were identified according to the different type of synthesis (Hydrothermal – HT, Microwave Irradiation – MW), pH (9 or 11) and synthesis time (6 or 12h, 1 or 36min), see Table 1.
Identification of the samples prepared according to the synthesis method, pH and employed synthesis time.
| Method | pH | Synthesis time | Code |
|---|---|---|---|
| Hydrothermal (HT) | 9 | 6h | HT9-6h |
| 12h | HT9-12h | ||
| 11 | 6h | HT11-6h | |
| 12h | HT11-12h | ||
| Microwave irradiation (MW) | 9 | 1min | MW9-1min |
| 36min | MW9-36min | ||
| 11 | 1min | MW11-1min | |
| 36min | MW11-36min | ||
The crystalline phases of the eggshell and hydroxyapatite powders were analyzed by X-ray diffraction technique (XRD, Bruker, D8 Advance), CuKα radiation (1.5404Å), 10–90° scan and step size of 0.02°s−1.
The crystallite size in hydroxyapatite powders was determined from the Scherrer Eq. (1):
where L is the crystallite size (nm), λ is the wavelength of the electromagnetic radiation used (0.154nm), β is the width at half height of the highest intensity diffraction peak and θ is the Bragg angle of the highest intensity diffracted peak.Fourier transform infrared spectroscopy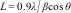
The functional groups present in hydroxyapatite were determined by Fourier transform infrared spectroscopy (FTIR, Shimadzu, IR-Prestige 21), between 400 and 4000cm−1.
Carbonate content in the materials was calculated from the FTIR results using the equation of Feathstone et al. [25]. For the calculation, the Ei extinction coefficients of the carbonate band present at or around 1458cm−1 and the phosphate band at or near 604cm−1 were obtained using Eqs. (2) and (3):
Here, T2 and T1 are the transmittance at the local baseline (1458 and 604cm−1) and the corresponding peak (1458 and 604cm−1), respectively.Dispersive energy X-ray spectroscopy-EDS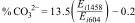
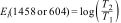
Semiquantitative chemical analysis of the material to obtain the Ca/P ratio was performed using dispersive energy X-ray spectroscopy (EDS, Oxford, Aztec Energy X-Act).
Transmission electron microscopyThe morphology of the powders was observed by transmission electron microscopy (TEM, Jeol, model JEM-2100) and the Image J software was used to calculate the particle size.
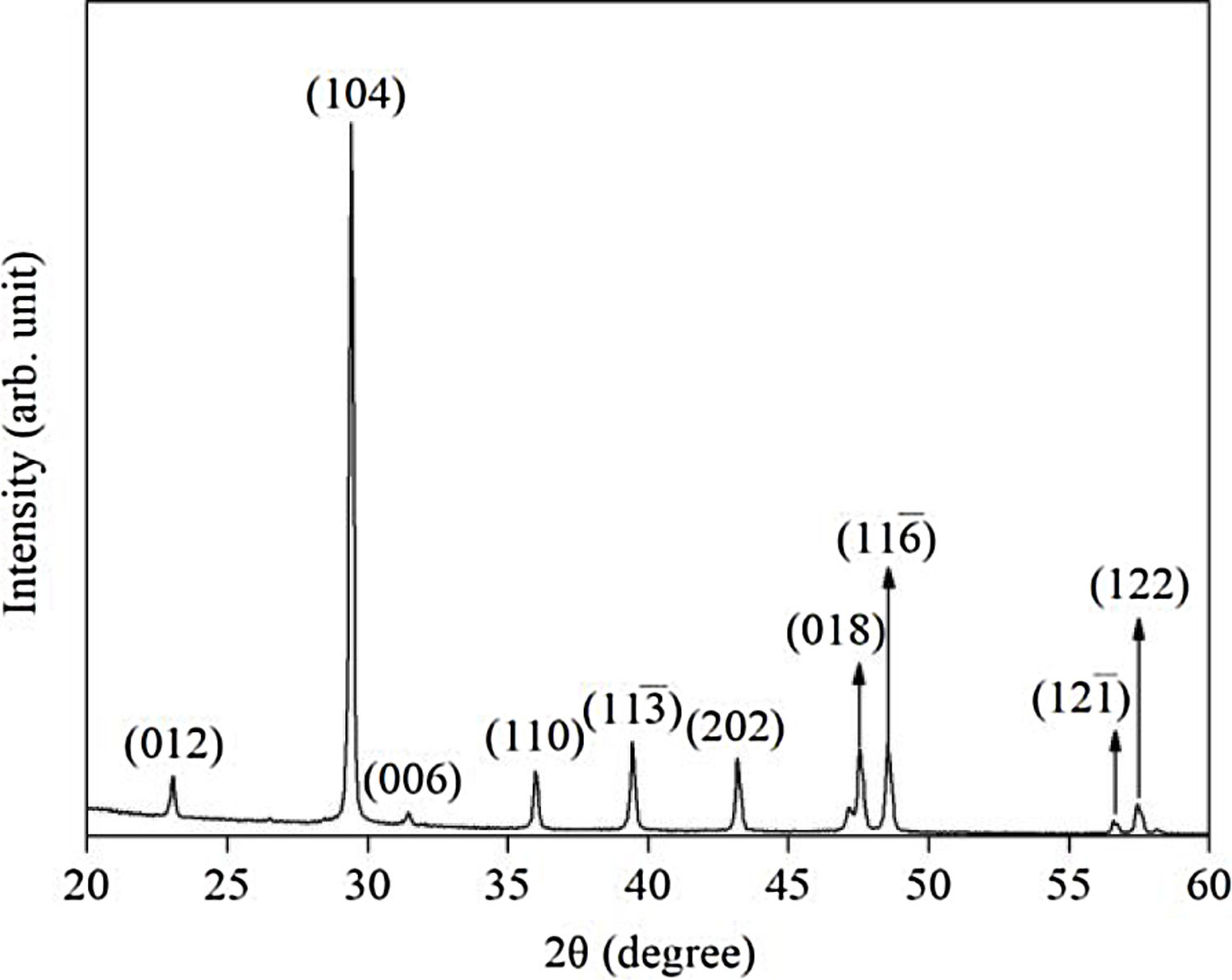
Results and discussionX-ray diffraction (XRD) of the eggshells and hydroxyapatite powdersThe XRD results from eggshells are shown in Fig. 1. The image displays the peaks corresponding to calcite (ICDD database 01-072-1937) with rhombohedrical crystalline system, proving in this way that the precursor material is mostly calcium, which are similar to previous researchers results [16,19,26].

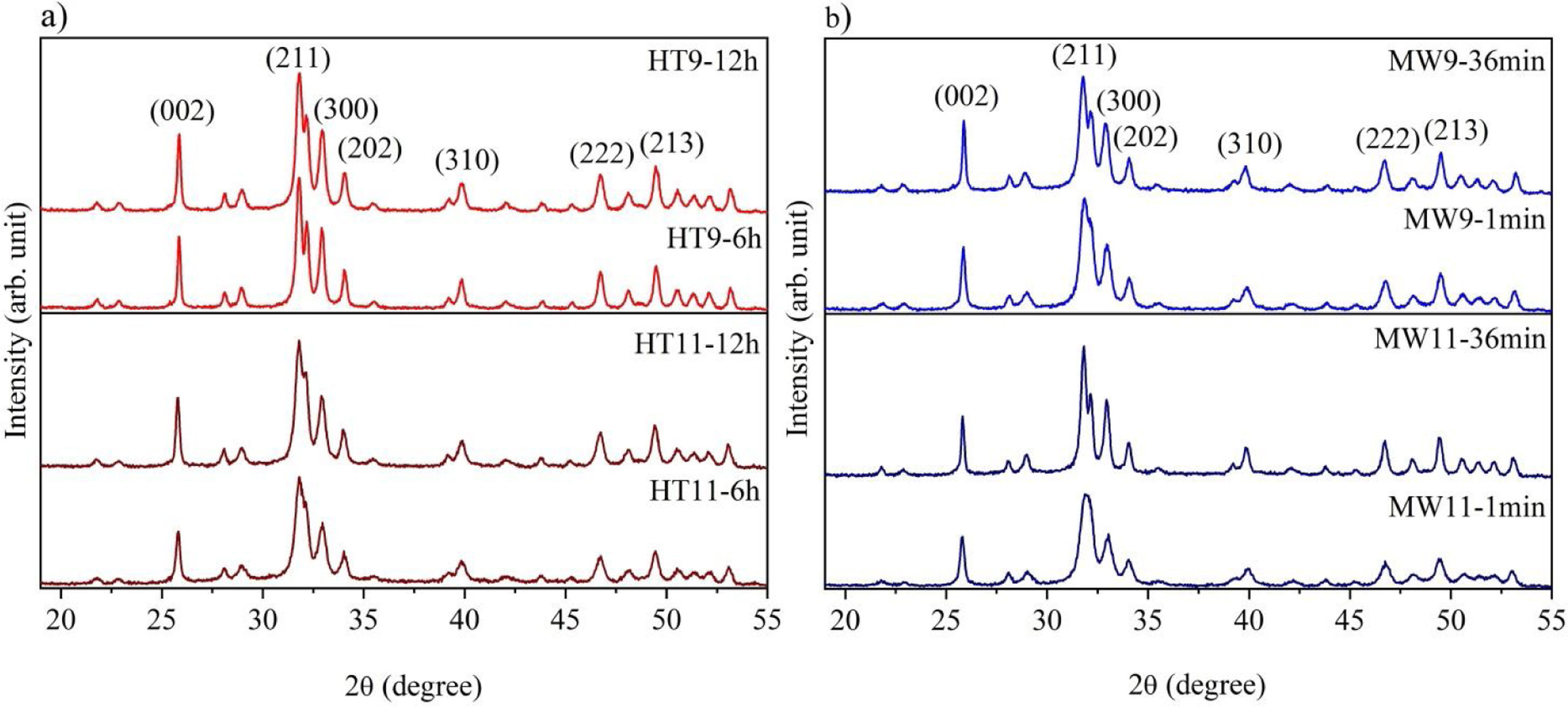
The XRD patterns of hydroxyapatite powders prepared by the two synthesis methods are shown in Fig. 2. It is noted that no other crystalline phase was detected besides hydroxyapatite, according to ICDD database 01-086-0740. This can be confirmed by the index peaks of Miller (002), (211), (300), (202), (310), (222) and (213) present in all sample standards and confirmed on the material crystallographic sheet [24]. For both employed techniques, the powders presented low crystallinity at the shortest synthesis times, as evidenced by the wide peaks of the XRD patterns of the samples [27,28]. However, crystallinity improved with synthesis time, which can be observed by increasing the intensity of the main peak (211) and decreasing peak width [29].
Fourier transform infrared spectroscopy hydrothermal and (b) microwave irradiation.")
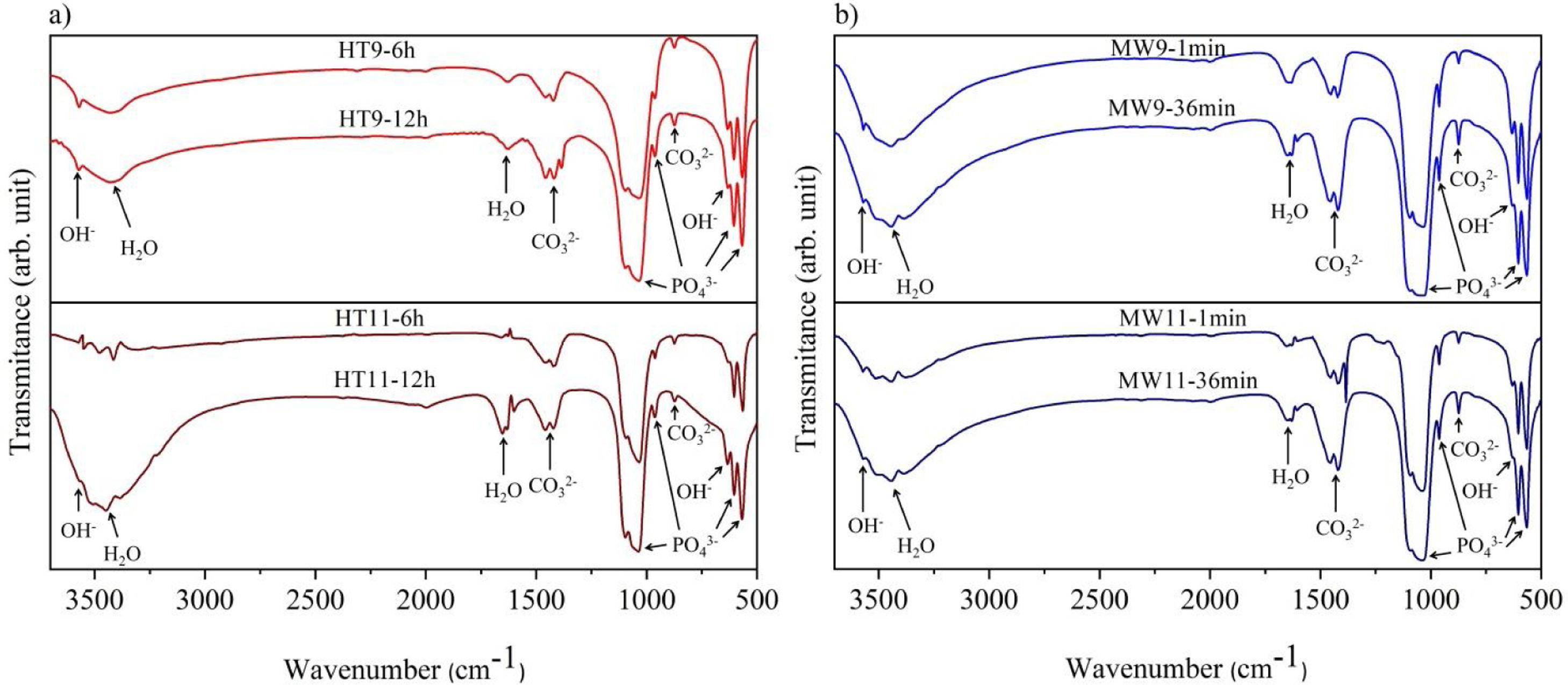
The vibrational spectra obtained by FTIR are depicted in Fig. 3. The bands in the region between 1033 and 1037cm−1 are assigned to the PO43− group, and are related to the asymmetrical stretching, v3, of the PO bonds and 962cm−1 to the symmetrical stretching, v1. An asymmetric flexion, v4, of the OPO bond is also observed in the regions of 565–566cm−1 and 603–604cm−1 respectively [22,30,31]. The OH− bands appear in the regions of 3567cm−1 and 671cm−1 refer to the symmetric stretching mode of the hydroxyapatite structural hydroxyl [32,33]. The vibrations of the adsorbed water molecules in the hydroxyapatite structure are present between 1629–1656cm−1 and 3430–3456cm−1[10]. The vibrations of the CO32− group appear in the regions between 1458–1460cm−1 and 874.5cm−1 show that hydroxyapatite is of Type B, i.e. when phosphate ions are replaced by carbonate ions absorbed from the ambient atmosphere during the synthesis of the material [30,34].
Crystal size, estimated carbonate values and Ca/P ratio hydrothermal and (b) microwave irradiation.")
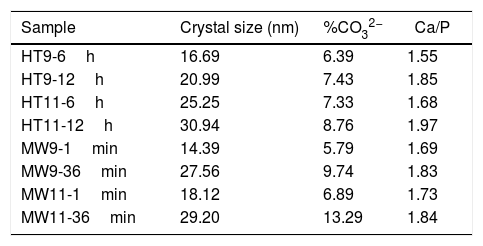
Table 2 shows the crystal size, the estimated carbonate values and the Ca/P ratio of all obtained samples. It is noted that the size of the crystal increased with the synthesis time. However, compared to bone hydroxyapatite crystallites, which are in the 20–40nm range, three samples with shorter synthesis times (HT9-6h, MW9-1min and MW11-1min) had smaller crystal size [35].
Results of crystal size calculation, carbonate percentage estimate and Ca/P ratio of hydroxyapatite samples.
| Sample | Crystal size (nm) | %CO32− | Ca/P |
|---|---|---|---|
| HT9-6h | 16.69 | 6.39 | 1.55 |
| HT9-12h | 20.99 | 7.43 | 1.85 |
| HT11-6h | 25.25 | 7.33 | 1.68 |
| HT11-12h | 30.94 | 8.76 | 1.97 |
| MW9-1min | 14.39 | 5.79 | 1.69 |
| MW9-36min | 27.56 | 9.74 | 1.83 |
| MW11-1min | 18.12 | 6.89 | 1.73 |
| MW11-36min | 29.20 | 13.29 | 1.84 |
From the estimated carbonate values, it can be seen that the HT11-12h, MW9-36min and MW11-36min samples exceeded the upper limit of the CO32− ion present in bone tissue hydroxyapatite, which is 8% [36]. This was due to the synthesis time which, when increased, favored the absorption of carbonate in the material, as well as the pH of the solution, since the solubility of CO2 increases in more basic media [37].
The closest samples to the theoretical Ca/P ratio of 1.67 were HT11-6h and MW9-1min. However, only the HT11-12h sample was above the Ca/P ratio of bone hydroxyapatite, which is between 1.5 and 1.85 [30,38].
Transmission electron microscopyThe TEM microstructures of the specimens selected by the two synthesis methods are shown in Fig. 4. For the hydrothermal method, it is possible to observe that well-defined nanorods were formed at both pHs. In the micrographs of the samples prepared by microwave irradiation there was also formation of nanorods, but much less defined and deformed.
 HT and (b) MW.")
The difference in the obtained morphologies by the two methods is related to the difference in the heating rate and synthesis time. This is due to in Hydrothermal synthesis the heating rate was slower and the synthesis time higher, which caused the hydroxyapatite particles to grow with more defined forms [39]. The Microwave Irradiation technique, because it has a faster heating rate, can cause rapid and volumetric heating during material processing, accelerating particle nucleation that will be predominant to the growth rate, thus affecting its morphology [40,41].
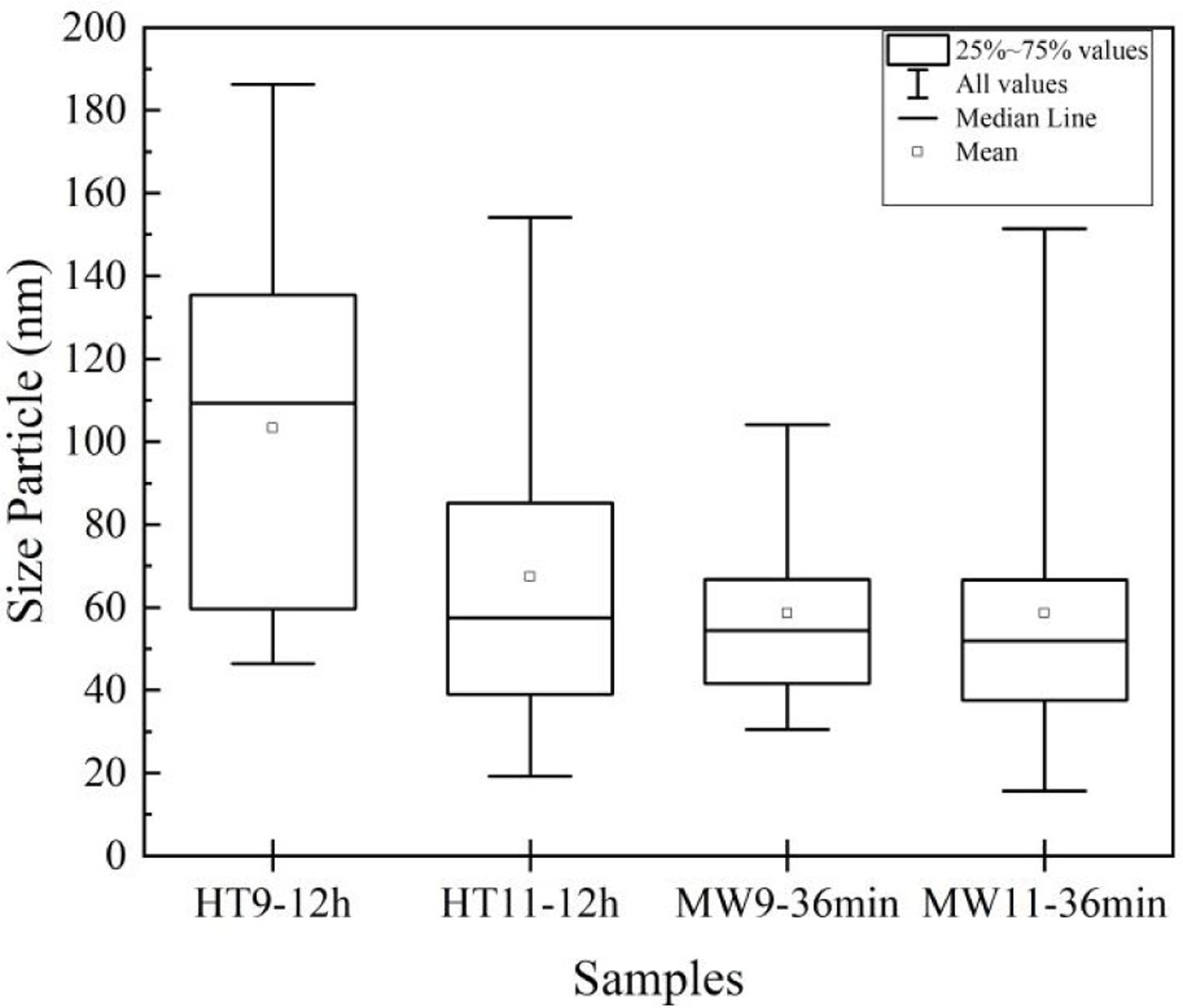
The average particle size results were analyzed by TEM and are shown in the graph of Fig. 5. For Hydrothermal samples, an average particle size of 30nm was obtained for the pH 9 and 50nm synthesized sample for the sample at pH 11, corroborating with Guang et al. [34] and Bucur et al. [42]. In the samples prepared by Microwave irradiation at pH 9, the particles had an average size of 40nm and at pH 11, 50nm. Those results are similar to the one obtained by Kumar et al. [30] and Apalangya et al. [43].

The nanorods morphology and a particle size between 20 and 80nm of the synthesized material in this work have same characteristics of bone hydroxyapatite, which favors its application as a biomaterial in orthopedics implants.
ConclusionNanometric Hydroxyapatite powders were obtained using two synthesis methods: Hydrothermal and Microwave Irradiation from chicken eggshell residue as a biogenic source.
For both methods the pH and synthesis time parameters were varied in order to find possible differences between the obtained products. XRD analysis showed that all powders obtained were homogeneous, with only hydroxyapatite as the single phase.
FTIR results indicated the presence of hydroxyapatite common organic groups. The Ca/P ratio indicated that only the HT11-12h sample was above the stoichiometric ratio of bone hydroxyapatite.
TEM analysis revealed the formation of rod-like particles with particle sizes similar to bone mineral. The synthesized material at pH 9 presented better results in relation to the absorption of carbonates, which is the most recommended for the preparation of hydroxyapatite.
For the synthesis method, as the Hydrothermal generated more well-defined particles, and similar to bone hydroxyapatite, this can be considered the ideal method for hydroxyapatite synthesis route and further application as a bone graft biomaterial.
The authors thank the financial support of the Brazilian research financing institutions: CAPES, FAPEMA and the Federal Institute of Maranhão (IFMA). The Materials Analytical Center for XRD analysis and Chemistry Analytical Center for the FTIR analysis at Federal University Maranhão (UFMA). The Analytical Physics Center from Federal University of Ceará (UFC) and the High-Resolution Microscopy Multi-User Laboratory at Federal University of Goiás.

