


Acute posterior multifocal placoid pigment epitheliopathy is an unusual, self-limited, inflammatory disease that affects the choriocapillaris, and external retinal layers.
Clinical caseA 26 year-old male patient complained of decreased visual acuity, as well as photopsia in both eyes for the past three days. Best corrected visual acuity was 20/200 in the right eye and 20/80 in the left eye. There was no anterior chamber inflammation or vitritis in either eye. There were plaque-like, cream-coloured sub-retinal lesions with ill-defined borders in the posterior pole of both eyes. Fluorescein angiography showed hypofluorescent lesions in early phases that corresponded to the lesions seen in the clinical examination. These lesions were hyperfluorescent in later phases of the angiography. Based on the clinical and angiographic findings, an acute posterior multifocal placoid pigment epitheliopathy diagnosis was made.
ConclusionsAcute posterior multifocal placoid pigment epitheliopathy is an inflammatory condition of unknown origin that is part of the differential diagnosis of placoid retinal diseases.
La epiteliopatía pigmentaria placoide multifocal posterior aguda es una entidad poco frecuente, autolimitada, de carácter inflamatorio a nivel de coriocapilaris y capas externas de la retina.
Caso clínicoPaciente varón de 26 años de edad, que acude por baja visual de inicio repentino en ambos ojos y 3 días de evolución, acompañada de fotopsias. A la exploración se encontró una capacidad visual de 20/200 en el ojo derecho y de 20/80 en el ojo izquierdo. No se encontró reacción inflamatoria en la cámara anterior ni tampoco vitritis en ningún ojo. En el polo posterior se encontraron lesiones de aspecto cremoso blanquecino subretinianas, en forma de placas. En la fluorangiografía retiniana se encontró hipofluorescencia en fases tempranas de las lesiones, vistas en fotos clínicas con hiperfluorescencia en fases tardías. En función de los datos clínicos y angiográficos se diagnosticó como epiteliopatía pigmentaria placoide multifocal posterior aguda.
ConclusionesLa epiteliopatía pigmentaria placoide multifocal posterior aguda es una entidad inflamatoria de origen desconocido que forma parte del diagnóstico diferencial, de las llamadas enfermedades placoideas de la retina.
Acute posterior multifocal placoid pigment epitheliopathy was described for the first time in 1968 by Gass.1 The report detailed the presence of multifocal placoid (plaque-like) lesions at the level of the external layers of the retina and the pigmented epithelium, in 3 otherwise healthy women. There is still debate as to the aetiology and precise location in the retinochoroid thickness of these lesions.2,3
In the majority of cases this condition is benign and self-limiting and does not require major intervention on the part of the medical provider, as in the cases initially reported by Gass.1,4
The clinical manifestations of acute posterior multifocal placoid pigment epitheliopathy present with sudden loss of central vision, which patients describe as: blurry vision, paracentral scotoma, metamorphopsia, “dots” in the visual field and photopsia.4
Seventy seven percent of cases present an initial vision of less or equal to 20/25, whereas in 58% of cases it is less or equal to 20/40.4
Visual deficiency can be uni- or bilateral (this is more common, and is present in 75% of cases).5 If the form of presentation is unilateral, the second eye occasionally becomes involved after a few days or weeks. It can be accompanied by ocular symptoms: headache, neck stiffness and occasionally general malaise, and with a history of a viral syndrome or recent vaccination.
In terms of epidemiology, both men and women are equally affected and it presents between the ages of 20 and 50.4,5
With regard to findings on in-depth examination of the eye, Gass1 described the presence of flat confluent cream-coloured lesions, with poorly defined margins dispersed throughout the posterior pole. The lesions appear behind the retinal equator and are bilateral. New lesions develop over several weeks after onset of symptoms, and therefore sometimes visible lesions appear at different stages. Occasionally they are associated with serous retinal detachment.6 This characteristic makes it difficult to distinguish acute posterior multifocal placoid pigment epitheliopathy from Harada's disease,7,8 and some authors have considered that these conditions might form part of the spectrum of the same disease.9
Although vitritis is not a significant component of acute posterior multifocal placoid pigment epitheliopathy, the presence and degree of intraocular inflammation widely varies. The following have been described: anterior uveitis, anterior granulomatous uveitis, and corneal stromal infiltrates.10
In general visual symptoms improve in 2–4 weeks, and therefore it is self-limiting.
Acute posterior multifocal placoid pigment epitheliopathy has a relatively good prognosis compared with other placoid white dot syndromes. However, Fiore et al.4 demonstrated that approximately 50% of patients completely recover their vision, therefore up to 25% of patients have 20/40 vision or worse. Sixty percent of the patients have residual visual symptoms.
In this sense, foveal involvement at the time of presentation is not a favourable prognostic factor, 88% of the eyes without foveal involvement completely recover vision, compared with 53% of the eyes with foveal involvement. Gass1 reported that visual recovery might continue for months after the lesions have been cured, even up to 6 months later.
The initial lesions at the level of the posterior pole eventually disappear, sometimes leaving areas of chorioretinal atrophy; only a few cases have an unfavourable result, as reported by Fiore et al.4 especially if these areas of atrophy involve the foveal centre.
Although it is one of the types of choriocapillaritis which are seen relatively often, there are still few reported cases of this disorder in national and international literature.
Clinical caseA twenty-six year old male patient, attending consultation reporting a three-day history of visual deficiency of sudden onset, accompanied by photopsies in both eyes. The patient did not provide a relevant history for the current disorder.
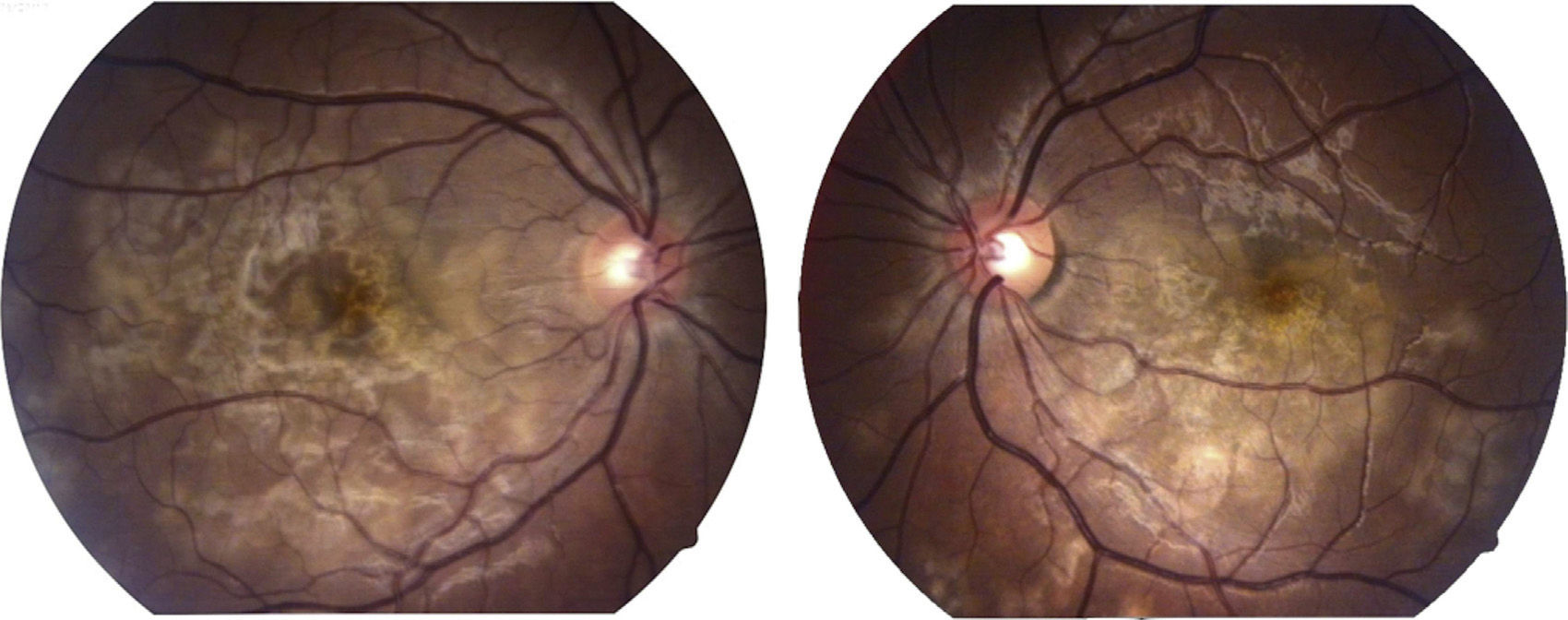
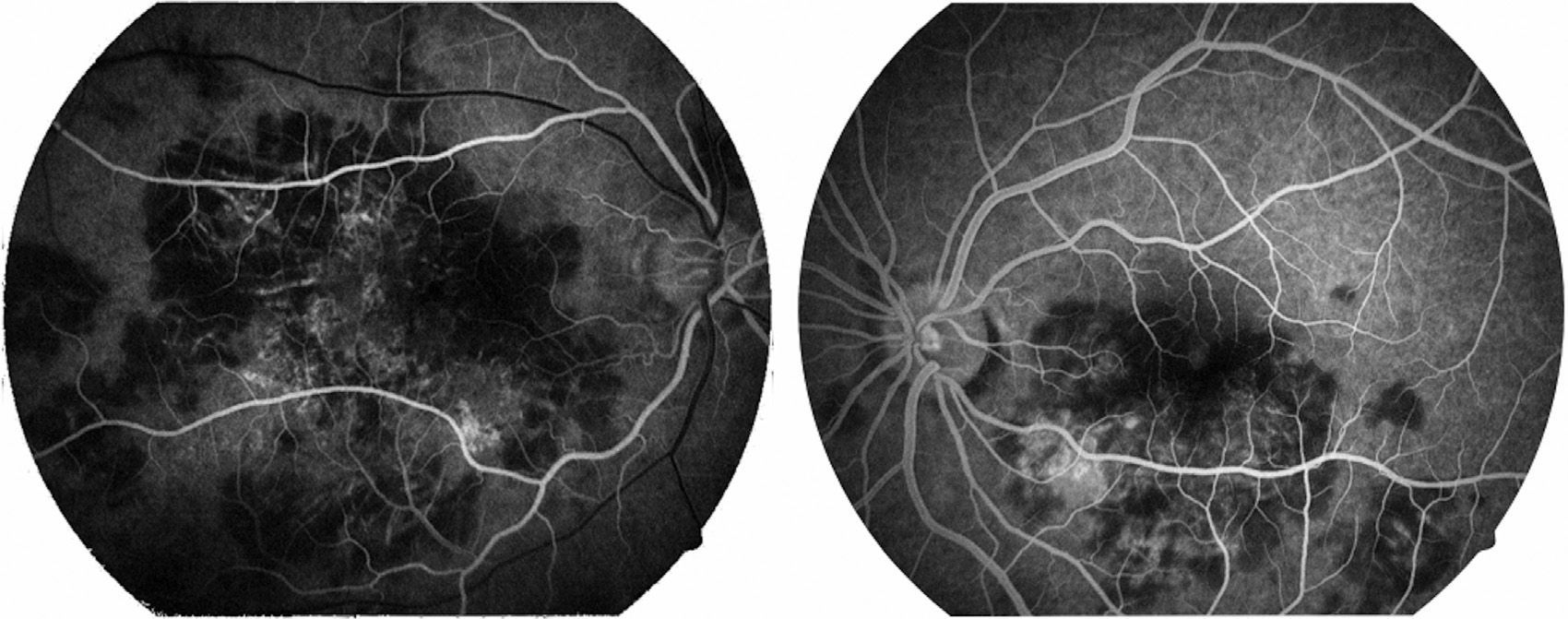
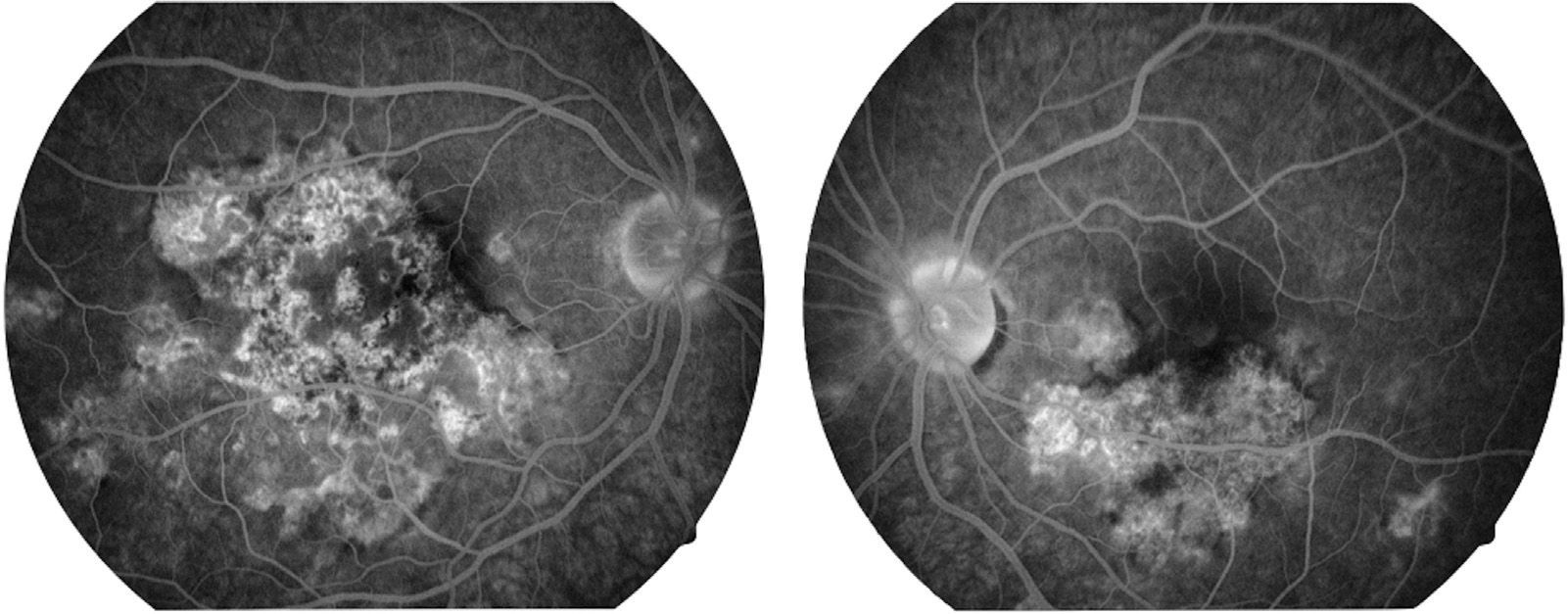
Visual capacity was 20/200 in the right eye and 20/80 in the left eye. No inflammatory reaction was found in the anterior chamber neither was the presence of vitritis. Limited creamy-white subretinal lesions were found in the posterior pole, more or less well defined, with a certain degree of confluence between them (Fig. 1). Retinal fluoroangiography showed hypofluorescence in the early phases which corresponded with the lesions seen in clinical photos in the posterior pole (Fig. 2), with hyperfluorescence of same in late phases of the study (Fig. 3).



Fluoroangiographic image in the late phase of both eyes. Hyperfluorescent areas can be seen in some of the areas which previously, in the early phases of the study, were hypofluorescent in both eyes. This change from early hypofluorescence to late hyperfluorescence of the lesions is characteristic of acute posterior multifocal placoid pigment epitheliopathy lesions.
From the clinical signs, the clinical image of the posterior pole of both eyes, as well as angiographs, a diagnosis was made of acute posterior multifocal placoid pigment epitheliopathy.
Symptoms started to improve and the lesions resolve, with a final visual capacity of 20/40 in the right eye and 20/25 in the left eye at 6 weeks’ follow-up without treatment.
DiscussionThe diagnosis of acute posterior multifocal placoid pigment epitheliopathy is principally clinical, and is based on examination of the posterior pole and on the evolution of symptoms over time. In the case of this patient, the diagnosis was confirmed by the visual deficiency, absence of other history, together with the presence of plaque-like lesions which were found by biomicroscopic examination with a Goldman 3-mirror lens, added to the findings of the fluorescein angiograph, where the presence of hypofluorescent areas in early phases of the study with late hyperfluorescence (which is characteristic of the disease) in areas corresponding to clinical lesions was seen, added to that which was self-limited. Furthermore, white dot syndromes, along with other conditions, come into the differential diagnosis of this disease; these include: serpiginous choroidopathy (which should be considered in recurrent, chronic cases), relentless placoid chorioretinitis, which should be taken into account in severe, persistent and recurrent cases (which was not the case with our patient), and evanescent white dot syndrome. Included in the differential diagnosis were: diffuse unilateral subacute neuroretinitis, Harada's disease, ocular tuberculosis, sarcoidosis, fungal disease, choroidal metastasis or lymphoid infiltrate, syphilis, vascular occlusions of the retina, central serous choroidopathy and rhegmatogenous detachment of the retina.
The cerebrospinal fluid should be tested if Harada's disease is suspected, in order to determine the presence of pleocytosis.
Retinal fluoroangiography is the study which best supports the diagnosis.
Gass1 described the early-phase lesions as hypofluorescent, and later on in the study there is progressive, irregular hyperfluorescent staining of the lesions (the aforementioned was a key sign in our patient's fluorangiographic study). As the process becomes inactive, hyperfluorescence can be seen corresponding to window defects in the pigmented epithelium of the retina, where the staining phenomenon is no longer evident.
Other types of studies performed are: indocyanine green angiography, optical coherence tomography, reporting abnormalities in the external layers of the retina.8 Furthermore, acute posterior multifocal placoid pigment epitheliopathy has been associated with central nervous system manifestations,11 which include: cerebral vasculitis, meningoencephalitis and cerebral vascular disease.
Headache is a common symptom and acute posterior multifocal placoid pigment epitheliopathy imitates migraine, including the aura.
Despite the fact that many patients report some history of viral disease, symptoms of general malaise and headache can be associated more with generalised underlying vasculitis. Systemic vasculitis12 has been involved in acute posterior multifocal placoid pigment epitheliopathy; and has also been associated with: erythema nodosum, ulcerating colitis, thyroiditis, nephritis, juvenile rheumatoid arthritis and granulomatous diseases such as: Wegener's granulomatosis (granulomatosis with polyangiitis) and pulmonary tuberculosis.
At present some people believe that a vascular lesion appears which affects the choroid, which might cause partial choroid ischaemia leading to damage to the pigmented epithelium of the retina, in the final instance affecting the photoreceptors. Imaging studies are compatible with this assertion. It is possible that a primary process involving the external retina and the pigmented epithelium of the retina might secondarily cause abnormalities of the choroid on angiography.
It seems that there might be a trigger effect, either inflammatory or infectious in nature which incites this process. Furthermore, associations with HLA-B7 and HLA-DR2 have been described, which suggests that some individuals are more susceptible. Although the exact aetiology is unknown, there might be an association with viral disease such as adenovirus infection.13 In half of the cases presented by Ryan and Maumenee2 there were prodromal manifestations of viral disease, and the association with bacterial infection is also described; in this process there is activation of the T lymphocytes sensitised by various stimuli such as: bacteria, virus and fungi. Activated T cells release lymphokines, which in turn activate macrophages and cytotoxic T cells. The macrophages then give rise to epithelioid cells and giant cells which can lead to the formation of granuloma.
Systemic associations, such as inflammation of the central nervous system, mentioned above, could be explained in this way. The presence of a thrombotic process of the choroid has also been postulated.
There is no specific treatment; immunosuppressants14 are used as well as intravenous steroids in patients with central nervous system involvement. Our patient was only monitored.
Choroid neovascularisations rarely develop for which intravitreal antiangiogenics are of use.
ConclusionsAcute posterior multifocal placoid pigment epitheliopathy is an inflammatory disorder, generally benign, rare, self-limited and which primarily affects the external layers of the retina, such as the choriocapillaris. Occasionally it involves the central nervous system, and must form part of the differential diagnosis of the inflammatory processes which affect the posterior pole, principally the so-called white dot syndromes.
It is important not to give unnecessary therapies, which far from helping the patient, might cause them further damage.
Conflict of interestsThe authors have no conflict of interest to declare.
The author thanks the staff of the Clínica David for their participation in preparing this case report.
Please cite this article as: Hernández-Da Mota SE. Epiteliopatía pigmentaria placoide multifocal posterior aguda. Reporte de caso. Cirugía y Cirujanos. 2016;84:133–137.