



Atypical haemolytic uraemic syndrome is one of the main variants of thrombotic microangiopathy, and is characterised by excessive complement activation in the microvasculature. It is also characterised by the clinical triad; non-immune haemolytic anaemia, thrombocytopenia, and acute renal failure. In addition, 60% of patients have mutations in the genes encoding complement regulators (factor H, factor I, membrane cofactor proteins, and thrombomodulin), activators (factor B and C3), as well as autoantibodies against factor H. Multiple factors are required for the disease to manifest itself, including a trigger and gene mutations with adequate penetration. Being one of the differential diagnoses of preeclampsia–eclampsia and HELLP syndrome means that the clinician must be familiar with the disease due to its high mortality, which can be modified with early diagnosis and comprehensive treatment.
El síndrome hemolítico urémico atípico es una variante de la microangiopatía trombótica, caracterizado por una excesiva activación del complemento. Se caracteriza por presentar anaemia hemolítica no autoinmune, trombocitopenia y falla renal aguda. Se ha observado que el 60% de los pacientes presentan mutaciones en los genes que codifican al complemento, tanto reguladores (factor H, factor I, cofactor de proteínas de membrana y trombomodulina), activadores (factor B y C3) como autoanticuerpos contra el factor H. Se requiere la presencia de múltiples factores para su manifestación, estos incluyen: un disparador y mutaciones de genes con la penetrancia adecuada. Es necesario que el clínico esté familiarizado con la enfermedad, ya que presenta una elevada morbimortalidad que puede ser modificada si se identifica de manera temprana y se da un tratamiento oportuno.
Atypical haemolytic uraemic syndrome is a variant of the thrombotic microangiopathy which is characterised by the following clinical triad: non-autoimmune haemolytic anaemia, thrombocytopenia and acute kidney failure.1,2
It is characterised by an irregularity of the complement system, caused by genetic mutation of its inhibitors.2 Malignant hypertension, septicaemia, auto-immune disorder (lupus, scleroderma), streptococcal infections, pregnancy, HELLP syndrome and cancer may be the cause of this syndrome.3
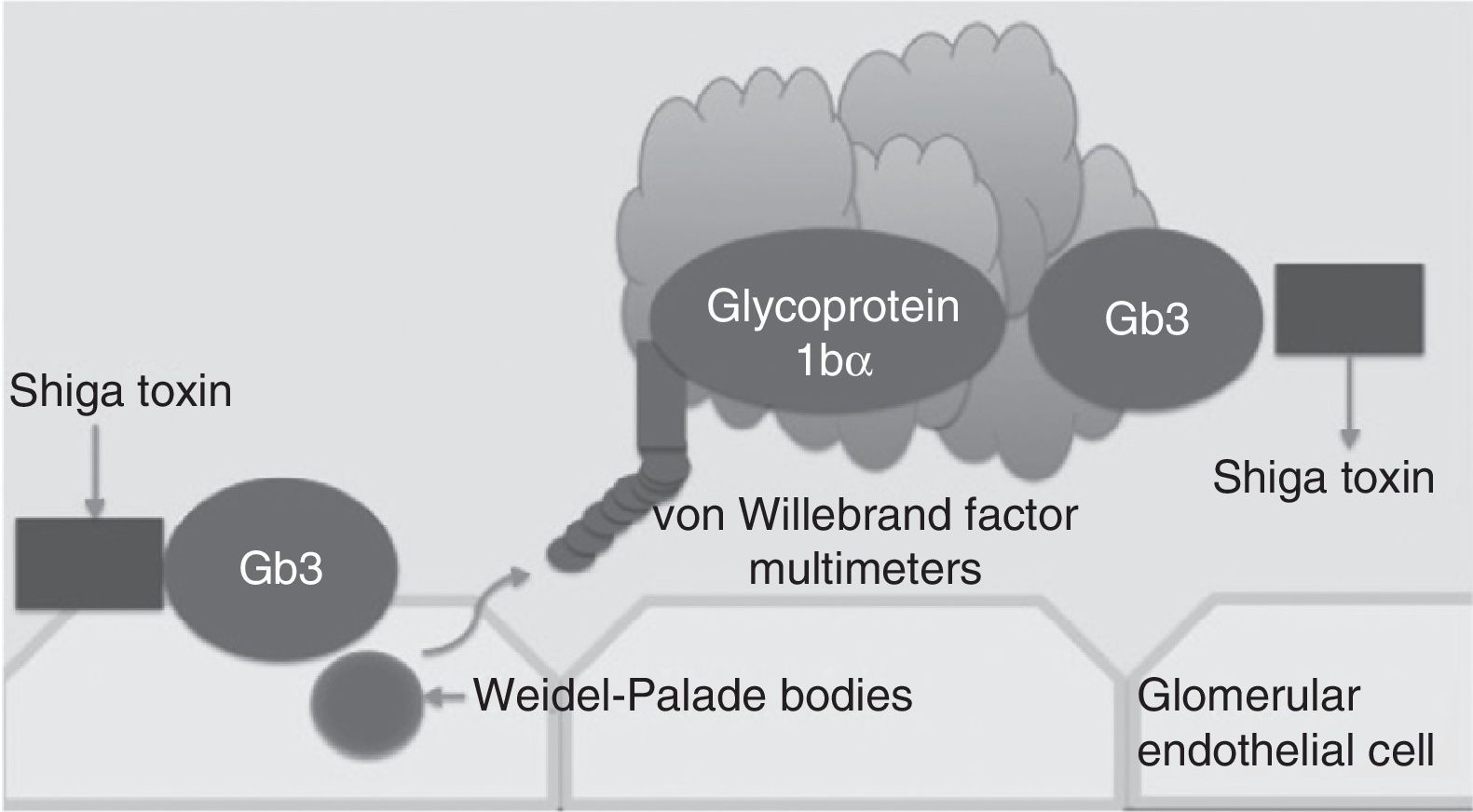
Traditionally, haemolytic uraemic syndrome is classified in 2 ways: typical haemolytic uraemic syndrome which peaks in incidence in children and is caused by enteric infections secondary to bacteria which are producers of the Shiga toxin (Fig. 1) and atypical haemolytic uraemic syndrome which in 50–60% of cases is associated with patients with gene mutations within the complement system, which in the majority of patients leads to terminal chronic kidney failure and the need for a kidney transplant.4–6

Pregnancy may be a trigger of this disease, particularly during the postnatal period. This is the result of the complement system playing a significant role in the physiopathology of pregnancy. It increases to prevent the damage caused by the placenta through the trophoblastic expression of complement regulatory proteins, known as the aggravating factor in degradation, the membrane cofactor protein (MCP) and CD59.5–7
There is a reduction of these proteins in the postnatal period, or a reduction in the majority complement proteins, which lead to the appearance of disease.7
EpidemiologyThrombotic microangiopathy associated with pregnancy (P-TMA) has an incidence of 1 case in every 250,000 pregnancies.8–10 There are reports of an incidence of atypical haemolytic uraemic syndrome of 2 cases for every 1,000,000 inhabitants.11 In women of reproductive age the phenotype of atypical haemolytic uraemic syndrome presents in late pregnancy or in the immediate postpartum period.11 In 10% of patients with this syndrome, the onset of pregnancy is the trigger.12
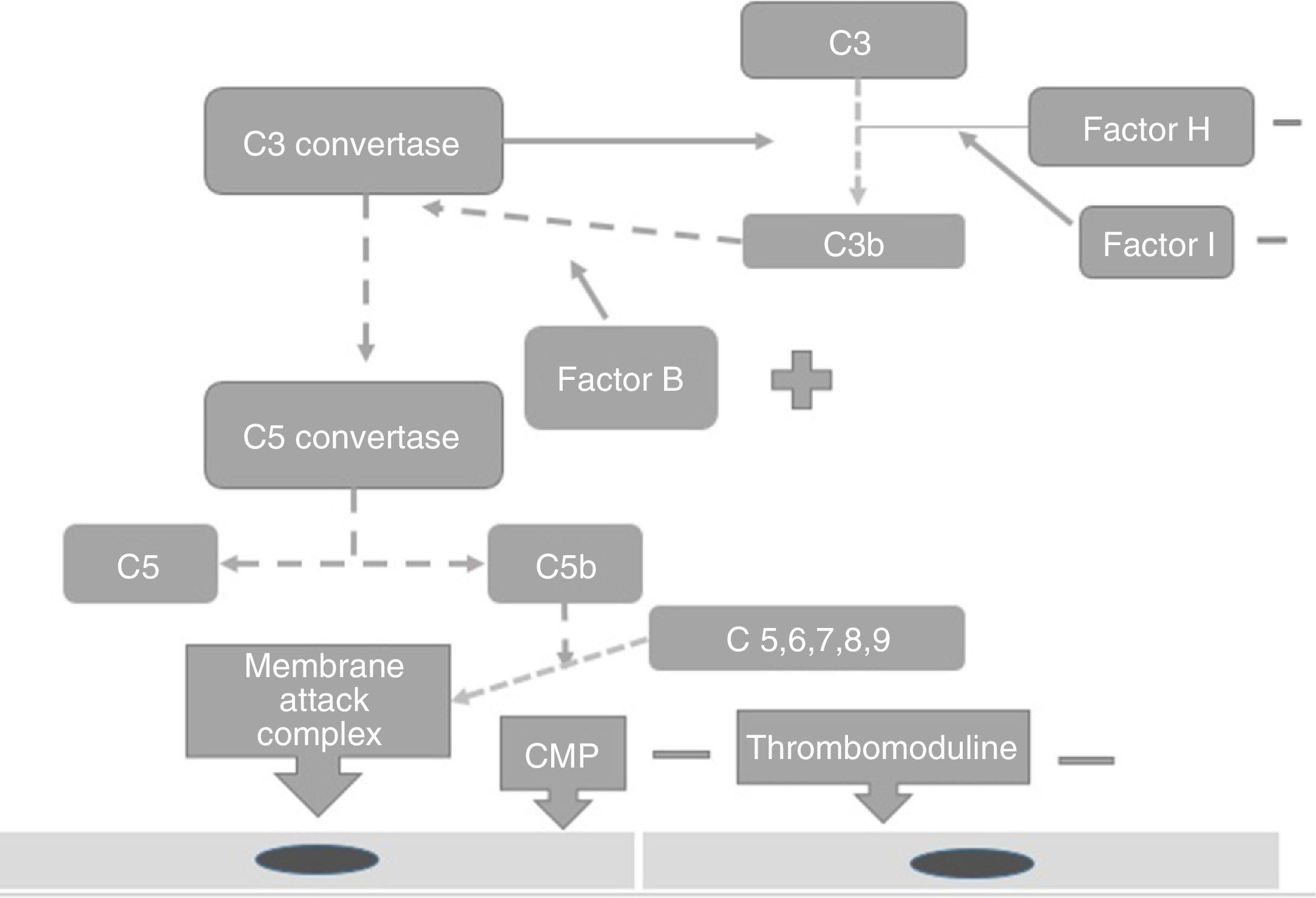
PhysiopathologyThe complement system is one of the main mechanisms involved in immunity measured by antibodies, and is considered to be a bridge between innate and adaptive immunity, which offers: protection against bacterial infection, enhancement of the elimination of immune complexes and inflammatory products, protection against external agents and the regulation of cellular apoptosis.13 There are 3 independent pathways for its activation: classical, lectin and alternative.14 Activation needs to be regulated to prevent tissue damage, particularly when activated by the alternative pathway.15 In the majority of patients affected by atypical haemolytic uraemic syndrome its development is related to uncontrolled activation of the complement pathway and with a growing number of genetic mutations which have already been identified for the activation of this syndrome.15 These mutations are observed in the genes which regulate the function of the complement, such as complement H (FCH) factor inhibitors. Richard16 identified the importance of the mutations of this gene as a cause of haemolytic uraemic syndrome. He discovered mutations of the FCH gene in exons 18–20 of 2 familial and 3 sporadic patients out of the 19 familial and 31 sporadic patients studied.15 Furthermore, this study showed that the familial haemolytic uraemic syndrome is a heterogeneous condition. Rodríguez17 discovered that the mutations in the FCH regulators, the complement I (FCH) factor and PCM led to the loss of the complement functioning, whilst those of C3 activated functioning (Fig. 2). PCM (CD46) is a transmembrane regulatory complement which is broadly expressed as FCH, and Richards18 discovered a PCM (CD46) mutation in individuals of 3 familials, who presented a deletion of 2 amino acids (D237/S238) in the familial 1 (heterozygote) and a replacement S206P, in the familial 2 (heterozygote) and 3 (homozygote), which confirmed that the deregulation of the complement predisposes the development of thrombotic microangiopathy and that patients under review for these defects could provide preventative treatment strategies in patients with this type of mutation.18 The FCH mutation occurs in 6–20% of patients with atypical haemolytic uraemic syndrome and the mutation in CD46 in 10% of patients.18–22

Factor I is an inhibitor of all complement pathways. It has the capacity to degrade C3b and C4b activated proteins in the present of cofactors such as FCH, CD46, among others. Incomplete deficiency of factor I is also associated with the development of atypical haemolytic uraemic syndrome.19
The increase in the function of the complement system factors, such as factors B and C3 may also cause the expression of this syndrome.20–22 Autoantibodies against FCH increase the risk for its expression but the disease has an associated incomplete penetration which is different to each mutation and therefore not all carriers of mutations will develop atypical haemolytic uraemic syndrome,22,23 only those women with a genetic predisposition and an external trigger.24
Atypical uraemic haemolygic syndrome and pregnancyThe pathogenesis of atypical haemolytic uraemic syndrome in pregnancy remains uncertain, it appears in 21% of adult women and in 79% of them it presents during the postnatal period.
The risk of presenting with gestational atypical haemolytic uraemic syndrome is greatest during second pregnancy. In a retrospective study Fakhouri25 found abnormalities of the complement system in 18 out of 21 patients. Findings did not differ among patients with pregnancy related or not related to atypical haemolytic uraemic syndrome. Mutations in the domain structures SCR19–20 of factor H were less frequent in patients with gestational atypical haemolytic uraemic syndrome compared with atypical haemolytic uraemic syndrome. Pregnancies with complement anomalies presented with complications characterised by foetal losses and preeclampsia in 4.8 and 7.7%, respectively.
Differential diagnosisMicroangiopathy may be the manifestation of many diseases: connective tissues diseases, cancer or may present in post transplant patients. However, it predominates in thrombotic thrombocytopenic purpura, atypical haemolytic uraemic syndrome, and haemolytic uraemic syndrome caused by the Shiga toxin from Escherichia coli. Thrombotic thrombocytopenic purpura is associated with a deficiency of ADAMTS13, and both types of uraemic syndrome are characterised by the presence of haemolytic anaemia, thrombocytopenia and multiple organ failure;26 atypical haemolytic uraemic syndrome, presents during the postnatal period whilst thrombotic thrombocytopenic purpura associated with deficiency of ADAMTS13 presents during pregnancy.26
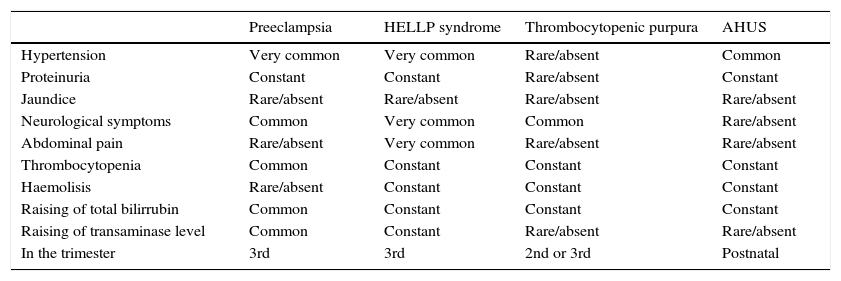
A key point for differentiating between atypical haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura is to highlight the presence of a severe deficiency of ADAMTS13, which should be greater than 5–10%.26–28 In cases where the activity of ADAMTS13 cannot be calculated, Coppo showed that the presence of a platelet count of 30×109/l and a creatine count below 2.26mg/dl is associated with deficiency of ADAMTS13 and hence with the diagnosis of thrombotic thrombocytopenic purpura (Table 1).29
Differential diagnosis of atypical haemolytic uraemic syndrome and other microangiopathies.
| Preeclampsia | HELLP syndrome | Thrombocytopenic purpura | AHUS | |
|---|---|---|---|---|
| Hypertension | Very common | Very common | Rare/absent | Common |
| Proteinuria | Constant | Constant | Rare/absent | Constant |
| Jaundice | Rare/absent | Rare/absent | Rare/absent | Rare/absent |
| Neurological symptoms | Common | Very common | Common | Rare/absent |
| Abdominal pain | Rare/absent | Very common | Rare/absent | Rare/absent |
| Thrombocytopenia | Common | Constant | Constant | Constant |
| Haemolisis | Rare/absent | Constant | Constant | Constant |
| Raising of total bilirrubin | Common | Constant | Constant | Constant |
| Raising of transaminase level | Common | Constant | Rare/absent | Rare/absent |
| In the trimester | 3rd | 3rd | 2nd or 3rd | Postnatal |
AHUS: atypical haemolytic uraemic syndrome.
The presence of acute kidney failure which requires replacement kidney transplant therapy is found to be associated with germinal mutations of the genes FCH, CD 46, FCI.30
Differential diagnosis of microangiopathy includes: disseminated intravascular coagulation, preeclampsia–eclampsia and HELLP syndrome.31
DiagnosisIn patients with suspected atypical haemolytic uraemic syndrome, the initial step is to confirm the presence of anaemia and thrombocytopenia and carry out differential diagnosis with diseases which may present these conditions. Initial laboratory studies should include a haematic biometry to document anaemia and thrombocytopenia, the presence of fragmented cells in blood smears, to determine the presence of a high lactate dehydrogenase as part of the haemolytic anaemia study, the raising of creatinine, the determination of ADAMTS 13, and a stool test and PCR detection for E. coli 0157.32
Diagnosis is complex since the majority of tests are not available in obstetrics services.31 In high risk patients or with genetic load for atypical haemolytic uraemic syndrome a DNA test is required to search for mutations in C3, FB, FH, FI, CD46. Formula testing in kinase diacylglycerol ¿ (DGK¿) is also proposed, as this encodes a protein which is not present in the complement system.32 The determination of these tests must be carried out prior to offering transfusion treatment to these patients.
Patients with complement-mediated atypical haemolytic uraemic syndrome present lower levels of C3 or C4. However, plasmatic levels within the C3, C4, FCB, FCH and TPI range do not exclude diagnosis of complement-mediated atypical haemolytic uraemic syndrome.33
TreatmentTreatment with plasmapheresis or infusion of fresh frozen plasma has been the cornerstone of treatment for atypical haemolytic uraemic syndrome since 1980 and was essentially the only available therapy until recently; the decision between one or the other form of therapy depends on the size of the patient and the extent of kidney failure, which limit the use of transfusion.3 Fresh frozen plasma infusion infuses normal functioning regulatory proteins from the alternative complement pathway, whilst the intended us of plasma exchange is to eliminate the following: dysfunctional proteins, anti-FCH antibodies and possible triggers of endothelial aggression (thrombogenic or inflammatory factors).4 In pregnancy initial treatment is plasmpheresis which is carried out in the same way as for non pregnant patients34, but prior to initiating therapy it is recommended that a viral panel be made to exclude any infection before transfusion, and vaccination is even recommended in susceptible patients.35 The most effective therapeutic regime is unknown, although guidelines of consensus recommend early initiation, to be maintained and intense, with daily session of plasmapheresis using 2 plasmatic volumes in adults and with a gradual reduction when the platelet count is above 150×109/l for at least 3 days, and concentrations of lactate dehydrogenase are normal. If no improvement is observed in a lapse of 5 days, the administration of eculizumab should be considered; if there is some improvement, the interval should be lengthened to once a week or every two weeks.35 If no plasmapheresis can be initiated in the first 24hours of presentation, fresh plasma transfusion will be initiated at a dose of 10–20ml/kg if the patient shows no signs of overload of volume or cardiac arrest, and the dose should be limited in cases of hypertension and kidney failure. Therapy duration and intervals between doses will depend on the clinical evolution of each patient. In some articles involving pregnant patients, treatment continues until pregnancy has run full term.35 Although giving birth in general will not resolve thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome, anecdotal evidence suggests that it may do so in some patients when they present with over-aggregated preeclampsia.36 If there is response to treatment, pregnancy should continue to full term with obstetric monitoring and interruption of pregnancy should be considered in the case of presentation of the alterations listed in Table 2.35–40
Indications for interruption of pregnancy in the atypical haemolytic uraemic syndrome.
| Haematological abnormalities |
| Thrombocytopenia or haemolytic microangiopathy which progresses despite treatment |
| Neurological abnormalities |
| Abnormal mental status (confusion, stupor, coma, disorientation) |
| Focal abnormalities (aphasia, dysarthria, focal motor deficiencies) |
| Renal abnormalities |
| Acute renal failure |
| Foetal abnormalities |
| Signs of distress or foetal suffering |
| Altered biophysical profile |
| Signs of restriction of intrauterine growth |
In patients with atypical haemolytic uraemic syndrome whose response to treatment is poor, administration of eculizumab should be considered. Its mechanism of action inhibits the complement pathway, specifically inhibiting C5 in the terminal complement pathway.
The use of eculizumab in gestational atypical haemolytic uraemic syndrome is anecdotal. In the last 10 years 7 articles have been published on its use, but there is not sufficient evidence for its recommendation as first line treatment. The dose reported in literature is 900mg weekly with intervals of 2–4 weeks, without any explanation of the cause of this difference between the doses and treatment continues with a maintenance dose of 1200mg every 7–14 days. They all report complete remission of the syndrome, without no recurrence in 6 months of follow-up.41–45 In patients with atypical haemolytic uraemic syndrome and replacement prolonged renal treatment, recuperation of kidney function is achieved with the use of eculizumab. There are reports of clinical remission of the disease which enables pregnancy to continue to 38 weeks with no repercussions to either foetus or mother.
PrognosisThis syndrome has a high rate of morbidity and mortality, but early diagnosis, correct monitoring and the right treatment improve prognosis. Patients in whom alterations in C3 have been reported require genetic advice owing to the risk of foetal loss and the development of preeclampsia. However, identification of this mutation does not provide any prediction of the risk of presenting with gestational atypical haemolytic uraemic syndrome, as the genetic penetration of this disease is variable. Published data report an overall penetration of 50–60% and additional factors, such as alterations in FH and PCM, which are the extra triggers for the development of atypical haemolytic uraemic syndrome. Any patient, therefore, with complement alteration must be strictly monitored by an obstetrician who is experienced in high risk pregnancy. Prophylactic plasmapheresis should not be carried out during pregnancy, when the patient has a background of atypical haemolytic uraemic syndrome, since the risk of complications exceeds the benefits of an empirical treatment.
ConclusionsThe progression and understanding of the physiopathology of atypical haemolytic uraemic syndrome during the last decade has made new treatment options possible. These may prevent its evolution in high risk patients. As pregnancy is a trigger of this syndrome, we believe it is necessary to assess high risk patients, which makes it necessary to be aware of the true prevalence of this disease during pregnancy. Studies are needed on the 2 current lines of treatment (plasmapheresis and eculizumab) to define the indications of both in patients with this syndrome and to know how effective they are in disease reversion as monotherapy or combined therapy.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Pérez-Calatayud ÁA, Briones-Garduño JC, del Pilar Álvarez-Goris M, Sánchez Zamora R, Torres Aguilar AA, Mendoza-Morales RE. Síndrome urémico hemolítico atípico en el embarazo. Cir Cir. 2016;84:344–349.