


von Willebrand disease is the most common inherited disorder of the coagulation proteins in humans. There are three types: 1, 2A, 2B, 2N, 2M and 3. It is associated with mutations on chromosome 12 in the region p13.2, encoding the von Willebrand factor (VWF), which is synthesised in endothelial cells and megakaryocytes.
DiscussionThe VWF gene has been characterised using molecular biology techniques, which have acquired an important role in diagnosis of von Willebrand disease, as well as in the investigation of alterations in other genes, which may be involved in regulating the synthesis, processing, and secretion of VWF. However, there are still no strategies to integrate the molecular biology diagnostic tests available.
Analysis of VWF multimers is a methodology that meets the characteristics for diagnosis, but it is not easy to standardise. Considering that even in tertiary centres in our country, von Willebrand patients do not have a definitive diagnosis, it is necessary to implement these methodologies to study and improve diagnosis.
Conclusionsvon Willebrand disease is highly heterogeneous due to the molecular mechanisms that produce the various clinical and laboratory phenotypes. In Mexico there are few studies related to this disease; therefore it is essential to conduct a comprehensive study including clinical, basic, and special testing laboratory tests, in order to establish a correct diagnosis, develop new therapeutic approaches, and offer the appropriate medical care and genetic counselling.
La enfermedad de von Willebrand es el trastorno hereditario más frecuente de las proteínas de la coagulación en los seres humanos. Existen 3 tipos: 1, 2A, 2B, 2N, 2M, y 3. Está asociada a mutaciones en el cromosoma 12, en la región p13.2, que codifica para el factor de von Willebrand (VWF), el cual se sintetiza en las células endoteliales y megacariocitos.
DiscusiónLa biología molecular ha permitido la caracterización del gen del VWF, adquiriendo un papel importante en el diagnóstico la enfermedad de von Willebrand así como en la investigación de alteraciones en otros genes, que pueden estar involucrados en la regulación de la síntesis, procesamiento y secreción del VWF. Sin embargo, aún no se han integrado las estrategias de biología molecular entre las pruebas de diagnóstico disponibles.
El análisis de los multímeros del VWF es una metodología que cumple con las características para el diagnóstico, pero no es fácil de estandarizar. Tomando en consideración que aún en los centros de tercer nivel en nuestro país los enfermos de von Willebrand no cuentan con un diagnóstico definitivo, es necesario implementar estas metodologías para su estudio y mejorar su diagnóstico.
ConclusionesLa enfermedad de von Willebrand es heterogénea debido a los mecanismos moleculares que producen los distintos fenotipos clínicos y de laboratorio. En México existen pocos trabajos relacionados con esta enfermedad, por ello es fundamental realizar un estudio integral que incluya aspectos clínicos, pruebas de laboratorio básicas y especiales, para establecer el diagnóstico correcto, desarrollar nuevos enfoques terapéuticos, y así ofrecer atención médica y asesoramiento genético adecuados.
Coagulation disorders are hereditary haemostatic abnormalities, and some may present considerable diagnosis and treatment difficulties. The von Willebrand disease is a hereditary disorder characterised by mucocutaneous haemorrhages of variable intensity, mainly affecting the primary haemostasis in platelet interaction, von Willebrand factor (VWF) and endothelium. It implies changes in the structure, function or concentration of the VWF, a plasmatic protein secreted by endothelial cells circulating in plasma.1
According to Vischer and de Moerloose, in 1926 Erik Adolf von Willebrand described a serious haemorrhagic disorder called pseudo haemophilia in a family with prolonged bleeding times despite having normal platelet counts.1,2 Later it was shown that this disorder is it related to a decrease in procoagulant activity in factor VIII of coagulation (FVIII), which may be compensated through an infusion of plasma or fractions of plasma, which evidences that the disease is caused by the lack of a plasma factor.3 In 1971, it was determined that FVIII and VWF were different proteins, and in 1975 Gralnick and Coller4 characterised the VWF. This discovery was accompanied by a new laboratory test using ristocetin to evaluate platelet function in this disease4,5 and, in 1985, the different nature of the VWF was definitely proved when describing the sequence of the VWF gene.5,6
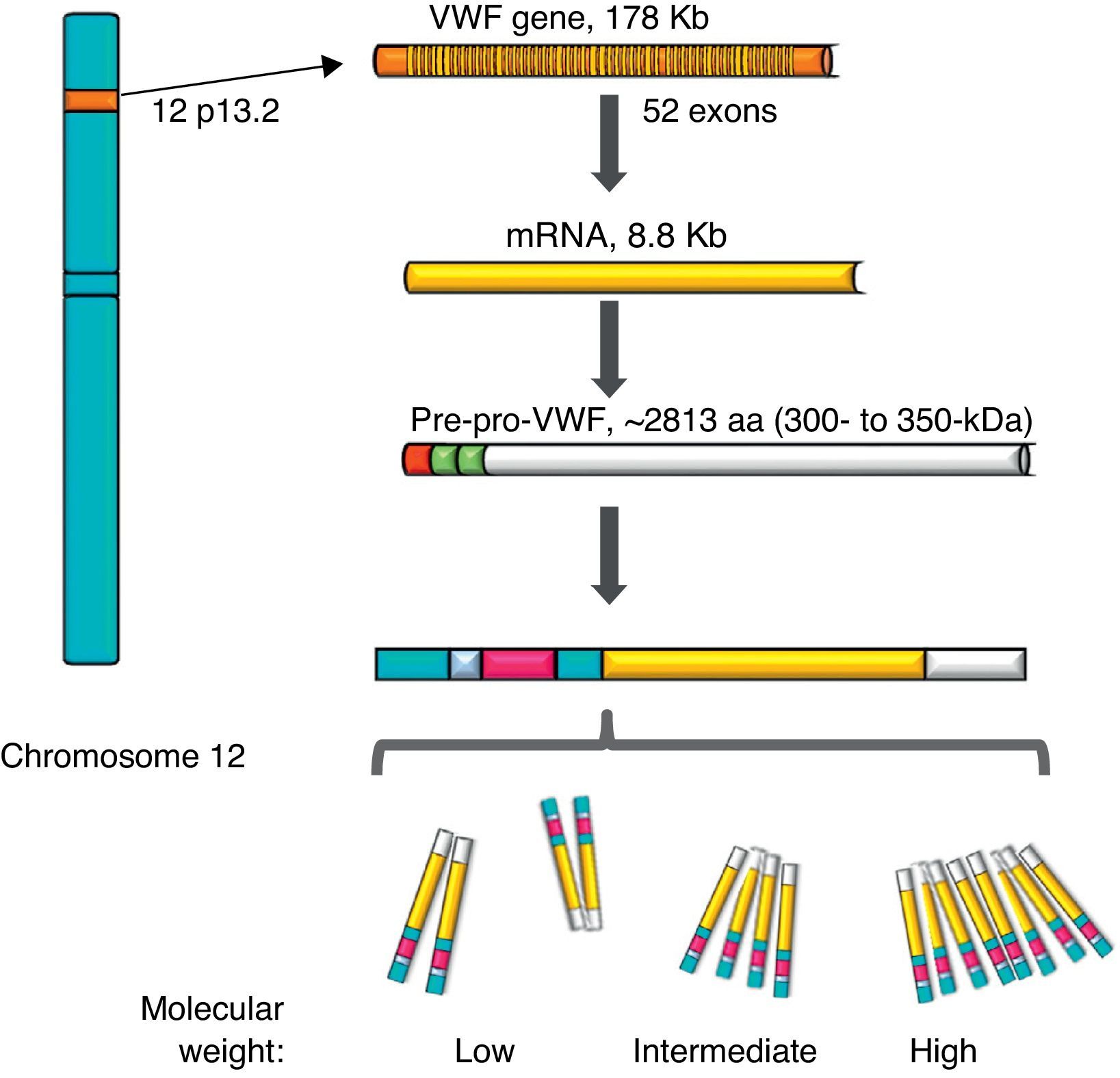
von Willebrand factorThe VWF is a multimeric protein synthetised in cells of the vascular endothelium, megakaryocytes and platelets with 12h average life, codified in a gene of 52 exons (178kb) localised in 12p13.2, and transcribes an mRNA of 8.8kb (2813 amino acids [aa]). Currently, more than 160 normal variants of the gene structure are known. Its transcription is regulated by specific cell-type transcription factors (GATA and ETS proteins), and there are also several repressive transcriptional elements in the ascending sequence of the gene (Fig. 1).7–9 On the other hand, there is a partial copy in chromosome 22 (pseudo gene) of exons 23–34 of the sequence of chromosome 12; this non-functional evolutional remnant shows a 3% sequence divergence in relation to the gene of chromosome 12 and seems to have been generated more or less during the time when major primates differentiated from monkeys, which complicates the related analysis.10 The VWF is a plasma glycoprotein (Gp); it is calculated that 75–85% of the VWF freely circulating in the plasma derives from the endothelium, whereas the remaining 15–25% is stored in circulating platelets which originate from the megakaryocyte.1,11

Representation of the gene, the transcription and the protein of the von Willebrand factor. The location of the von Willebrand factor gene is observed in chromosome 12, the mature mRNA and the processing of the protein, from the pre-pro-von Willebrand factor, to the formation of multimers with high molecular weight.
During the synthesis of the VWF the pre-pro-VWF protein is formed: it is an initial product of 300–350kDa (∼2813 aa), which contains a signal peptide of 22 aa, a propeptide of 741 aa and a mature protein of 2050 aa. The structure of the protein of the VWF is formed by several repeated domains in the order D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. D1, D2, D′ and D3 domains participate in the regulation of formation of multimers, and the D′ and D3 regions also mediate the union with the FVIII. Both the A1 and the A3 domains have collagen-joining properties. The areas where the VWF joins the platelets are: in the A1 domain to the Gp Ib/IX platelet receptor (GpIb/IX), and in the C2 domain to the Gp IIb/IIIa receptor (GpIIb/IIIa), in such a way that each VWF monomer has domains which allow the protein to join platelets ligands (GpIb/IX and GpIIb/IIIa), in the subendothelium (collagen) and in the bloodstream (FVIII).7 The protein carries out a post-translational incorporation, its signal sequences are acknowledged by the Sec62/63m complex associated to the translocon in the membrane of the endoplasmic reticulum. The propeptide of 2791 aa initiates the process of formation of the protein of the VWF and forms dimers through disulphide bridges in carboxi-terminal positions, through the disulfide isomerase membrane. Later, glycosylation takes place and is transported to the Golgi apparatus, where the mature protein of 2051 aa forms disulphide bridges in the amino-terminal portions of the dimers, and consequently, series of multimers of different sizes ranging from one fundamental 225kDa unit to 120,000kDa (Figs. 1 and 2).11,12
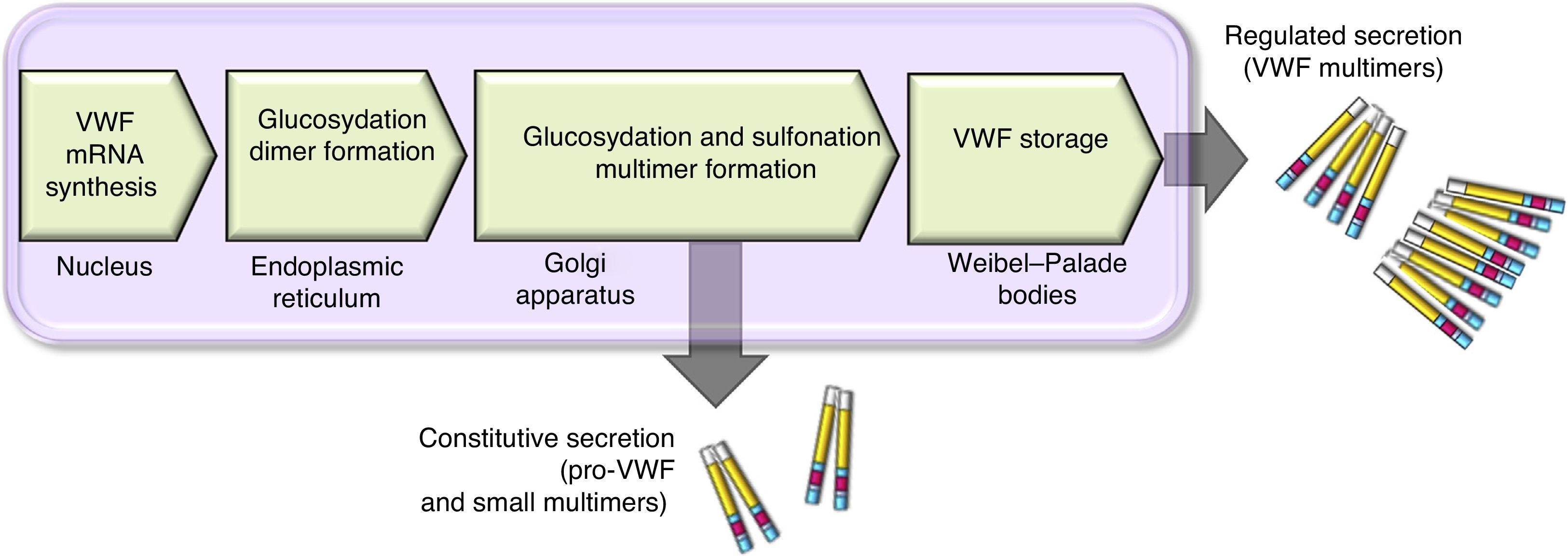
There are 2 paths involved in the secretion of the VWF. The constitutive path is related to the synthesis of plasma VWF of endothelial origin stored in the Weibel-Palade bodies and the regulated path, which involves the release of VWF fully multimerised, stored in the alpha granules, in megakaryocytes and platelets (Fig. 2).13,14
, where maturation ends forming multimers. A small amount of immature VWF, dimers and small multimers are constitutively released.")
Scheme of the processing and secretion of the von Willebrand factor in endothelium cells. The VWF is formed in the endoplasmic reticule, where glycosyl is dimmed. Dimers are transported to the Golgi apparatus and continue in the secretion granules (Weibel-Palade bodies), where maturation ends forming multimers. A small amount of immature VWF, dimers and small multimers are constitutively released.
The VWF shares these storage areas with other proteins released in response to a variety of physiological and pharmacological stimuli, including thrombin, shear stress and desmopressin.15 Currently it is known that after its release from the cell where it is synthetised, multimers of extremely large and high molecular weight VWF join the endothelial cell surface through interaction with the P-selectin protein of the Weibel-Palade bodies. In this area, VWF multimers are subject to the shear stress of the bloodstream, and a physiological reduction of the size of multimers through controlled proteolytic fragmentation, by the ADMTS-13 metalloproteinase acting on multimers of the VWF in the A2 domain, between aa 1605 and 1606. This process originates the different forms of the factor, ranging from simple dimers, to the 20 units forming the most complex multimer. Proteolytic degradation of multimers is a normal event, since these have an elevated thrombogenic potential, through the areas of interaction with platelets, and the wall of blood vessels. Under homeostasis conditions this reaction inhibits the growth of the thrombus forming the platelets.16,17
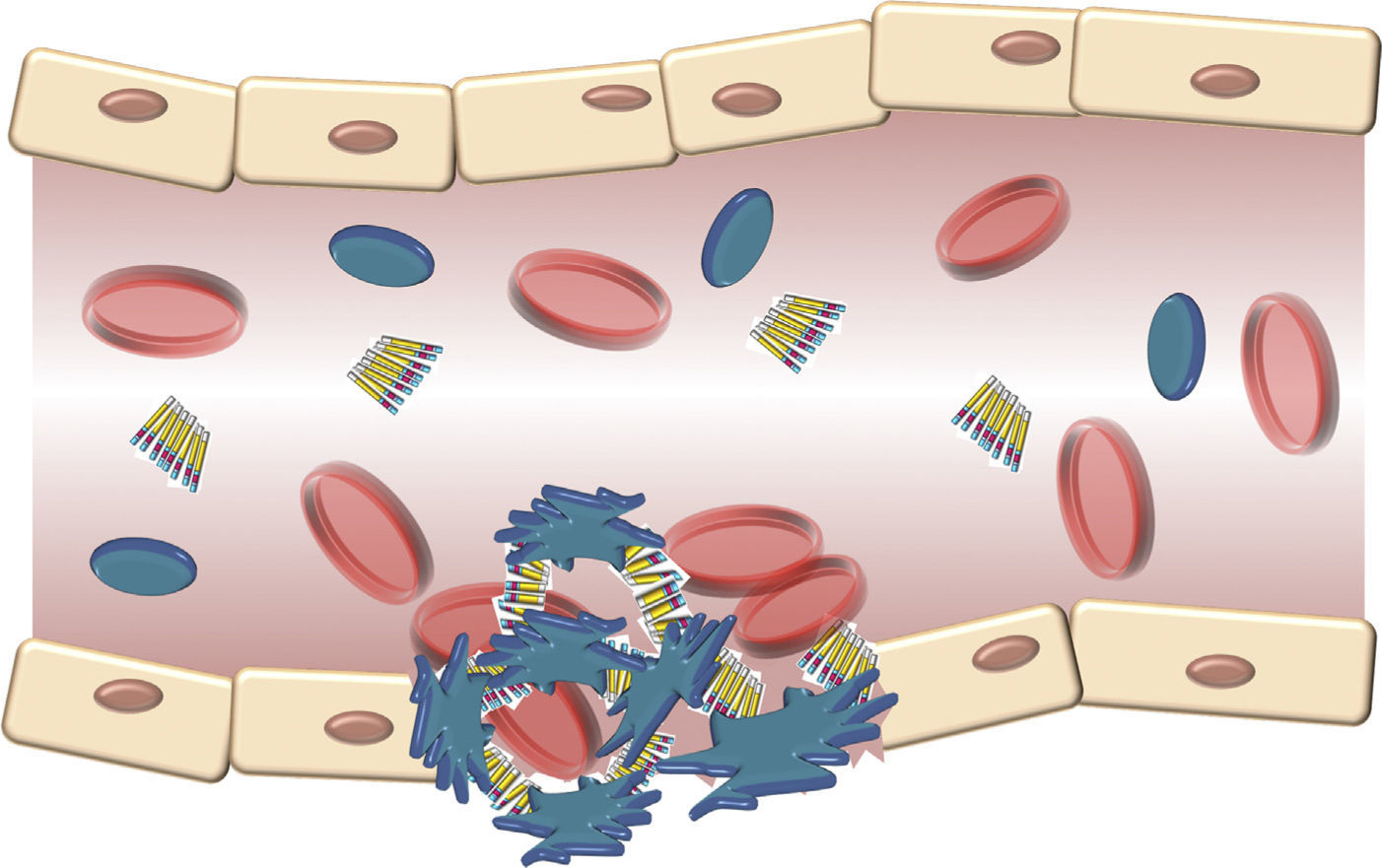
The structure of the VWF indicates that its main role is to join various ligands in the bloodstream and the blood of the damaged vessel (Fig. 3). Therefore, the 3 physiological functions of the protein are: (A) mediating in the adhesion of platelets to the areas of vascular damage when joining the GpIb/IX platelet receptor, and collagen in the vascular subendothelium; (B) facilitating platelet aggregation by joining the GpIIb/IIIa platelet receptor; and (C) joining the FVIII and protecting it from proteolytic degradation caused by C protein activated in the bloodstream.16 Finally, these contacts reach a threshold leading to platelet activation. Then, platelets adhere in a stable manner to the wall of the damaged vessel and experience a response of aggregation through an event mediated by a platelet receptor, the GpIIb/IIIa.17

Schematic description of the interaction of the von Willebrand factor and platelets activated during the formation of the platelet cap in the haemostasis. Platelets adhere in a transitory manner to the von Willebrand factor, and their function is acting as a bridge between the GpIb/IX receptor in the surface of the platelets and the collagen fibrils of the subendothelium.
The von Willebrand disease is the most common hereditary coagulation disorder in humans, presenting global distribution, and is also common in other animal species such as dogs and pigs.18 Its prevalence varies, depending on the approach to define the diagnosis. In at least 2 major prospective studies it was found that 1% of the predominantly paediatric population presents symptoms and laboratory signs of von Willebrand disease.19,20 It is believed that the prevalence of von Willebrand disease with haemorrhagic symptoms is approximately 1 in 1000.21 In Mexico there is no epidemiological record of the disease; only a few studies have been reported from the clinical and haematological standpoint, and recently one pilot study in 133 patients with suspected von Willebrand disease.22
General information on the von Willebrand diseaseThe von Willebrand disease is transmitted in an autosomal dominant way; it is the most common hereditary haemorrhagic disorder. It is caused by the decrease in the amount of VWF or the presence of a qualitatively abnormal VWF in circulation. Rarely, the von Willebrand disease may be an acquired disorder. The manifestations of the disease are mucosal cutaneous bleedings (epistaxis, bleeding gums, bleeding of other mucosa, ecchymosis, bleeding in dental procedures, etc.). Its diagnosis depends entirely on laboratory coagulation tests and their classification demands special studies (such as determination of the multimers of the VWF). It is important to classify the type, since this is important in choosing the treatment. Most of the time the treatment is with desmopressin, and only in the most severe cases is the factor replaced.23
Clinical diagnosisA true, detailed and complete clinical history will allow the doctor to make an approximation regarding the type of alteration of the existing haemostasis. It is necessary to bear in mind that, while a positive family history helps in clarifying the diagnosis, a negative family history does not exclude the possibility of a congenital haemorrhagic anomaly. Family history may even sometimes fail to provide conclusive evidence. Family history in patients with von Willebrand disease is often unclear; it must be suspected in people with excessive mucocutaneous haemorrhages, such as haematomas with no acknowledge traumas, spontaneous onset, or minimum traumas; also, haemarthrosis is always abnormal and indicates the existence of a serious coagulopathy. The same can be said about spontaneous muscle haematomas. They are both an essential symptom of haemophilia, and its onset is very rare in other haemorrhagic diathesis, except in severe von Willebrand disease, especially type 3 and likewise, prolonged recurrent nose haemorrhages and buccal cavity haemorrhages, including gum bleeding after brushing teeth or flossing, or prolonged bleeding after spot grinding or extractions. There may even be haematuria, excessive or prolonged bleeding after a surgery or trauma, and affected women also usually experience menorrhagia (generally from menarche), and prolonged or excessive bleeding after delivery. It is also important to take into consideration several other factors, some of which increase the concentration of the VWF: pregnancy, exercise, trauma, surgery, age; and its level is higher in people with blood type A than in blood type O. On the other hand, other factors cause their decrease: hyperthyroidism, renal failure, liver disease, atherosclerosis, inflammatory conditions, cancer, diabetes and hypothyroidism.24,25
Haematological chemical laboratory testsThe haematological chemical laboratory provides very useful information that must be interpreted in relation with the clinical context, which provides a greater chance of a definite diagnosis and proper treatment. Tests are divided into basic, which allows us to have a general idea of the condition of the patient, which include: blood cytometry, bleeding time, prothrombin time, partially activated thromboplastin time, and thrombin time; afterwards, tests studying primary, secondary haemostasis and fibrinolysis, as well as tests targeted to the diagnosis of thrombotic problems.26,27
Basic testsBasic tests are platelets count, bleeding time, prothrombin time, partially activated thromboplastin time and thrombin time. The first 2 explore primary haemostasis, while the remaining 3 evaluate secondary haemostasis. If the results are normal, except in rare cases haemostatic disorders of clinical meaning are ruled out.27,28 Its lengthening may be due to deficiency of a factor or the existence of an antibody, inhibitor of coagulation proteins. The correction, or not, of the test, when mixing the patient's plasma with normal plasma, will show if it is a deficiency or an inhibitor.29
Specific testsSpecific tests allow quantification of individual factors. They are used when one or more basic tests are altered, or when, while being normal, there is clinical suspicion of a systemic coagulopathy. There are 3 types: trial, functional and immunological. The first type, in turn, may be coagulometric methods using plasmas with a deficiency in the factor to be analysed.
Immunological methods, through antibodies, assess the concentration of the antigen of the protein (but not its function). A discrepancy between the levels obtained through functional methods and immunological methods reveals the existence of molecular anomalies.
Within specific tests there is the activity of VWF as cofactor of ristocetin (VWF:RCo), which depends on the presence of large multimers, antigenic VWF (VWF:Ag), FVIII coagulant (FVIII:C), ristocetin-induced platelet aggregation (RIPA); most subtypes have a decrease in the response to ristocetin, which is an agonist of the receptor of GpIb-IX and the VWF. Also, we can include the joining capacity of VWF to collagen and the FVIII:C (VWF:CB and VWF:VIII) and the analysis of platelet function (PFA-100®). The latter may substitute the bleeding time in the future, the use of which is very controversial.
Analysis of the multimers of the von Willebrand factorThe analysis of the multimers of the VWF, based on their molecular weight, is made through electrophoresis. Multimers are seen using an autoradiogram after the incubation with antibodies against the VWF marked with immunoperoxidase or alkaline phosphatase. This technique allows a definite diagnosis of the different types of von Willebrand disease based on its multimers pattern, and takes place only in the laboratories referred to.24,25,30,31
Classification of the von Willebrand diseaseThe last time the International Society of Thrombosis and Haemostasis (ISTH) published its recommendations regarding the classification of the von Willebrand disease was in 2006, which allowed that disease to be subdivided as quantitative deficiencies (types 1 and 3) or qualitative deficiencies (type 2), based mainly on the phenotype of the protein of the VWF.25,32
Type 1 von Willebrand diseaseThis is the most common form of von Willebrand disease and represents nearly 80% of all cases. Its transmission is autosomal dominant, with incomplete penetration. It is characterised by a mild to moderate reduction (0.45–0.05U/ml) in plasma levels of VWF:Ag and VWF:RCo. The VWF is normal from the functional standpoint, like the range of multimers of plasma VWF, while the plasma level of FVIII:C is reduced in proportion to the level of VWF. These patients show a spectrum of mucocutaneous haemorrhagic symptoms, the severity of which, in general, is correlated with the level of deficiency of the VWF. Molecular studies indicate that, in addition to the mutations in the gene of the VWF and the ABO blood group, alterations in other genes may influence the mild decrease of the VWF, such as those involved in the regulation of plasma levels of the VWF and the FVIII.33 Although there seem to be many different mutations causing the type 1 disease, at least in one, the mutation originating in the replacement of tyrosine with cysteine in the 1584 codon, it is found in 10–20% of patients in the US and Europe.25,32,34
Summary for some type 1 results. Below is a summary of some of the expected laboratory results for type 1: decreased levels of FVIII, VWF:Ag and VWF:RCo; decreased or normal levels of FVIII:C and ristocetin (RIPA). VWF:RCo/VWF:Ag>0.6. Normal levels of multimers, but with partial quantitative deficiency. Frequency: 70–80% and autosomal dominant or co-dominant inheritance.25,32–34
Type 3 von Willebrand diseaseType 3 von Willebrand disease has a prevalence of 1–3 per million in most populations, although in some places where marriage to blood relatives is common, the prevalence is considerably higher. The disorder is autosomal recessive and most parents of patients with type 3 disease show few, or no, haemorrhagic symptoms; however, in individuals affected, it is the most severe form of the disease.
In the type 3 disease, the levels of VWF:Ag and VWF:RCo are always <0.05 U/ml, and frequently undetectable. The plasma level of FVIII:C is reduced to levels from 0.01 to 0.10U/ml. In general there are no plasma multimers. These patients show serious recurrent mucocutaneous haemorrhages, as well as frequent musculoskeletal and soft tissue haemorrhages. With the passing of time, if the treatment is not correct, there is chronic musculoskeletal damage and patients of mean age could require joint replacement surgery.25,32–34
Summary of expected laboratory results for type 3. Markedly low levels of FVIII, VWF:Ag and FVIII:C. Slightly low levels of VWF:RCo. Absent ristocetin (RIPA) and multimers. Quantitative variant with decrease of FVIII levels. Frequency: 1–5/106 and autosomal recessive.
Type 2 von Willebrand diseaseThe current classification of the von Willebrand disease acknowledges 4 different qualitative forms of the disease: subtypes 2A, 2B, 2M, and 2N.25,32–34
Type 2A von Willebrand diseaseThis condition is characterised by loss of the platelet-dependent function of the VWF due to the absence of forms of the protein with high molecular weight. It exists either because of a biosynthetic incapacity to produce these multimers or because these are produced, segregated and subsequently degraded prematurely in the plasma. The typical characteristic of the type 2A disease is a relationship between the VWF:RCo and the VWF:Ag (<0.6), with an absence of VWF multimers with high molecular weight and affect on the platelet agglutination capacity induced by the RIPA. Substitute mutations causing the type 2A disease have been located in D2, D3, A1, A2 domains and terminal C of the VWF gene.25,32–34
Summary for some results of type 2A. Lower levels of FVIII, VWF:Ag, VWF:RCo and RIPA. Lower or normal levels of FVIII:C. VWF:RCo/VWF:Ag>0.6. Absent multimers with high molecular weight, and lower levels of those with intermediate molecular weight (qualitative deficiency). Frequency: 10–15% and codominant inheritance, rarely recessive.
Type 2B von Willebrand diseaseThis subtype of the von Willebrand disease represents a classic genetic feature of function gain. The disorder is the result of a variety of dominant substitution mutations in the joining region of the GpIb in the A1 domain of the VWF. These mutations increase the adherence capacity of the VWF to this platelet receptor and produce spontaneous interactions between VWF and the platelets in the bloodstream, a phenomenon that does not take place with normal VWF. In the blood samples, this interaction may be observed as accumulation of the platelets, and this same abnormality frequently produces mild chronic thrombocytopoenia. In other tests, the relation between VWF:RCo and VWF:Ag will often be <0.6, with a deficit of multimers of VWF with high molecular weight in the plasma because these have joined the platelets. Another important test to confirm the presence of the type 2B disease is the proof of increase in the platelet agglutination capacity induced by the RIPA; it is detected by platelet aggregation to low concentrations of ristocetin <0.6 mg/ml.
This set of clinical and laboratory findings may also be observed in a rare hereditary platelet disorder, the pseudo von Willebrand disease or platelet type von Willebrand disease. In this dominant hereditary feature, the substitution mutations of function gain are in the gene of the Ib platelet GP and cause an increase in the affinity of adhesion to the A1 domain of the VWF. To differentiate it from type 2B of the platelet type von Willebrand disease, tests of agglutination induced by ristocetin to washed platelets of the patient and mixed with normal plasma are necessary (which will show a greater reactivity in the case of platelet type von Willebrand disease, but not in type 2B), or the analysis of the genes of the VWF and of the GpIb.25,32–34
Results for type 2B. Decreased or normal levels of FVIII and FVIII:C. Slightly decreased levels of VWF:Ag. Decreased levels of VWF:RCo. RCo/VWF:Ag <0.6. Increased levels of RIPA. Absent or low levels of multimers with high molecular weight, described as a qualitative variant with increase of affinity to platelets by the GpIb/IX complex, a frequency <5% and codominant inheritance.
Type 2M von Willebrand diseaseThis subtype is characterised by the loss of function, equivalent to type 2B of the disease. Most substitution mutations causing the type 2M of the von Willebrand disease have also been located in the A1 domain of the VWF. In the type 2M disease, the relation between the VWF:RCo and the VWF:Ag is also <0.6, but the characteristics differentiating it from type 2B include the absence of accumulation of platelets (and therefore thrombocytopenia) and the presence of a normal pattern of multimers in the plasma.25,32–34
Results for type 2M. Decreased or normal levels of FVIII and FVIII:C. Slightly decreased levels of VWF:Ag. Decreased levels of VWF:RCo. RCo/VWF:Ag <0.6. Decreased or normal levels of RIPA. Multimers present. It is a qualitative variant with decrease of platelet function, rarely presents and with a codominant inheritance.
Type 2N von Willebrand diseaseThis last qualitative mutating form of the von Willebrand disease is different from all the previous ones in several aspects. Its inheritance pattern is autosomal recessive, and frequently the only laboratory abnormality is a reduced plasma level of FVIII (generally from 0.10 to 0.40U/ml). Type 2N von Willebrand disease is one of the differential diagnoses of an isolated level, from low to moderately low in FVIII.33–37
Patients may be homozygotes for substitution mutations or compound heterozygous for 2 different mutations. They may also have mutations in the joining area or a null mutation. Nearly 20 mutations have been described. Most are located between exons 18 and 20, which affect the domain of joining to FVIII. Other mutations have been described in exons 17 and 21–27, which are outside the area of joining to FVIII, and are also responsible for the decrease in the joining capacity between VWF and FVIII. The R854Q mutation is the most frequent mutation reported.
Results for type 2N. Decreased levels of FVIII and FVIII:C. Slightly decreased or normal levels of VWF:Ag. Normal levels of VWF:RCo and RIPA. Multimers present. It is a qualitative variant with decrease of the joining to FVIII, very rare and with an autosomal recessive inheritance (Table 1).
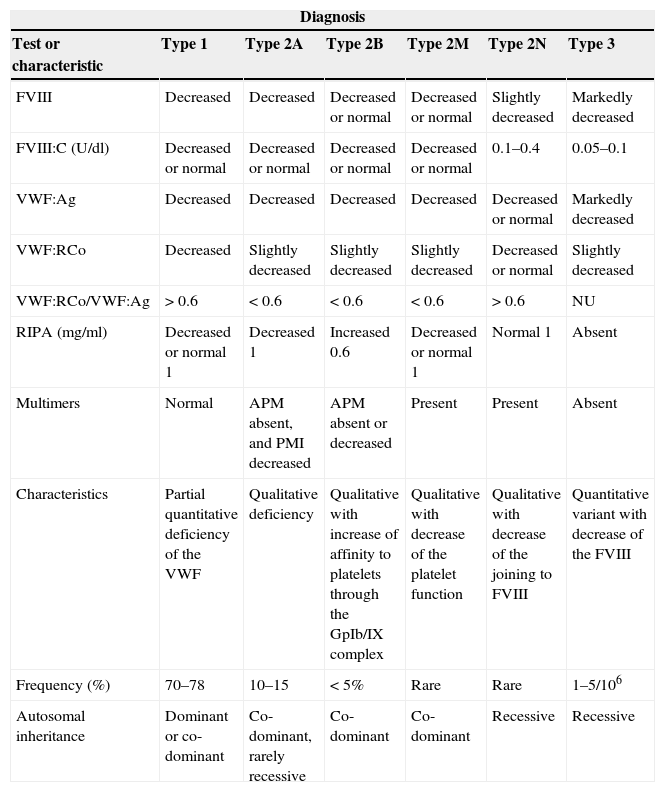
Haematological chemical tests and inheritance of the types of the von Willebrand disease.
| Diagnosis | ||||||
|---|---|---|---|---|---|---|
| Test or characteristic | Type 1 | Type 2A | Type 2B | Type 2M | Type 2N | Type 3 |
| FVIII | Decreased | Decreased | Decreased or normal | Decreased or normal | Slightly decreased | Markedly decreased |
| FVIII:C (U/dl) | Decreased or normal | Decreased or normal | Decreased or normal | Decreased or normal | 0.1–0.4 | 0.05–0.1 |
| VWF:Ag | Decreased | Decreased | Decreased | Decreased | Decreased or normal | Markedly decreased |
| VWF:RCo | Decreased | Slightly decreased | Slightly decreased | Slightly decreased | Decreased or normal | Slightly decreased |
| VWF:RCo/VWF:Ag | >0.6 | <0.6 | <0.6 | <0.6 | >0.6 | NU |
| RIPA (mg/ml) | Decreased or normal 1 | Decreased 1 | Increased 0.6 | Decreased or normal 1 | Normal 1 | Absent |
| Multimers | Normal | APM absent, and PMI decreased | APM absent or decreased | Present | Present | Absent |
| Characteristics | Partial quantitative deficiency of the VWF | Qualitative deficiency | Qualitative with increase of affinity to platelets through the GpIb/IX complex | Qualitative with decrease of the platelet function | Qualitative with decrease of the joining to FVIII | Quantitative variant with decrease of the FVIII |
| Frequency (%) | 70–78 | 10–15 | <5% | Rare | Rare | 1–5/106 |
| Autosomal inheritance | Dominant or co-dominant | Co-dominant, rarely recessive | Co-dominant | Co-dominant | Recessive | Recessive |
APM: multimers with high molecular weight; FVIII: factor VIII of coagulation; FVIII:C: factor VIII coagulant; NU: not useful; PMI: multimers with intermediate molecular weight; RIPA: platelet aggregation induced by ristocetin; VWF: von Willebrand factor; VWF:Ag: VWF antigenic; VWF:RCo: VWF as cofactor of ristocetin.
Allelic variants in the genome of the VWF imply effects on the phenotype, harmful, neutral or beneficial. Normal variants are very common, and around 150 have been described to date, such as the c.1451A>G variant causing a change of a histidine for an arginine in position 484 of exon 13, which are related to the different populations studied.38,39
There is a great variety of factors involved in the origin of pathological variations, due to changes in only one nucleotide, mainly in the D3 domain (Fig. 1), such as the “Vicenza” variant (c.3614G>A) in exon 27, which causes a change of an arginine with a histidine in position 1205 of the VWF and thus decreases the time that the VWF remains in the plasma; this mutation is one of the most studied mutations and is related with type 1 von Willebrand disease.40,41 There is even a molecular study of the VWF gene in Mexican mixed-race patients, where 3 new mutations are described: E1447Q in a patient with type 1 von Willebrand disease and diabetes; P2781S in a patient with type 2M; and P812L in another patient with type 1/2N.22,42 This high degree of polymorphisms in the VWF, together with the great size of the gene and the presence of a partial pseudo gene, make the complete sequencing of the gene and interpretation of data difficult and complex. These variations and their frequencies can be widely consulted in the database of the International Society on Thrombosis and Haemostasis-Scientific and Standardisation Committee ISTH-SSC VWF Online (VWFdb).
As has been described, the great number of variations on the gene, and with them their effects on the structure and function of the VWF, cause different forms of von Willebrand disease. In addition, other diseases may be related to quantitative and qualitative defects in the VWF. New evidence provides precise information on the mechanisms of the disease and the risk of bleeding associated with the deficiency or abnormality of the VWF.43
TreatmentThe treatment depends on the subtype of the von Willebrand disease; however, therapies to prevent or control bleeding recommend: increasing plasma concentration of VWF by endogenous release through the stimulation of endothelium cells with desmopressin; likewise, transfusion therapy with blood products; also using agents promoting haemostasis and healing of wounds, but not altering plasma concentration of VWF substantially.
Therapeutic decisions depend on the type and seriousness of the von Willebrand disease, as well as the seriousness of the bleeding, and its actual or potential nature. The treatment is variable and frequently based on the local experience and the doctor's preference. There are standard recommendations to guide the treatment of von Willebrand disease.44,45
DiscussionMolecular biology has allowed the characterisation of the gene of the VWF, as well as determining allelic variants and mutations. One of the most important applications of molecular biology is aimed at molecular diagnosis, which is supported by basic or clinical chemical laboratory analysis tests, special tests and genetics. Molecular biology tests play an increasingly important role in the diagnosis of the von Willebrand disease, greatly improving the capacity to characterise the genetic variants of the disease. Molecular biology tests are also used in the investigation of other genes which may be involved in the regulation of the synthesis, processing, secretion and control of plasma levels of VWF. Likewise, given the seriousness of the phenotype of the type 3 von Willebrand disease, the genetic prenatal diagnosis of this variant offers results to families and their doctors to make informed decisions regarding family planning. However, given the complexity of the full study of the gene, it is hard to search for a specific mutation, which is why molecular biology strategies have not yet been integrated into the available diagnosis tests. On the other hand, analysis of the multimers of the VWF is a methodology with the necessary characteristics for diagnosis, although it is technically demanding and not easy to standardise. Taking into consideration that in third level centres in our country, patients with von Willebrand disease do not have a conclusive diagnosis, it is necessary to implement these methodologies to study and improve diagnosis.
ConclusionThe von Willebrand disease is very heterogeneous, due to the molecular mechanisms producing the different clinical and laboratory phenotypes. In Mexico there are few works related to this disease, and therefore it is essential to carry out a comprehensive study including clinical aspects, basic and special laboratory tests on this type of patient in our population, to establish a proper diagnosis and develop new therapeutic approaches for treatment, to prevent and/or correct the related alterations, and offer correct medical care and genetic advice.
Conflict of interestThe authors declare that there are no conflicts of interest.
Please cite this article as: Hernández-Zamora E, Zavala-Hernández C, Quintana-González S, Reyes-Maldonado E. Enfermedad de von Willebrand, biología molecular y diagnóstico. Cir Cir. 2015;83:255–64.