




La irrupción de la lipoproteína (a) (Lp(a)) en la valoración de los factores de riesgo cardiovascular es quizás, junto con el descubrimiento y uso de los fármacos inhibidores de la proproteína convertasa subtilisina/kexina tipo 9 (iPCSK9), la mayor novedad en el campo desde hace décadas. La concentración de Lp(a) (especialmente los niveles muy elevados) tiene una innegable asociación con determinadas complicaciones cardiovasculares, como los derivados de enfermedad vascular aterosclerótica (EVA) y la estenosis aórtica. Sin embargo, existen varias limitaciones actuales tanto para establecer asociaciones epidemiológicas como para realizar un tratamiento farmacológico específico. En primer lugar, la medición de la Lp(a) depende en gran medida del test utilizado, principalmente por las características de la molécula. En segundo lugar, la concentración de Lp(a) está determinada en más del 80% por la genética, por lo que, al contrario de otros factores de riesgo cardiovascular no puede ser modificada con cambios del estilo de vida. Finalmente, aunque existen múltiples ensayos clínicos prometedores con fármacos específicos para reducir la Lp(a), actualmente solo los iPCSK9 (limitados para su uso por su coste y eficacia limitada) reducen de forma significativa la Lp(a).
Sin embargo, y en línea con otras sociedades científicas, la Sociedad Española de Arteriosclerosis (SEA) considera oportuno con el objetivo de aumentar el conocimiento sobre la contribución de la Lp(a) al riesgo cardiovascular, la elaboración de un documento donde se recoja el estado actual del tema, las recomendaciones de control del riesgo cardiovascular global en las personas con Lp(a) elevada y sus implicaciones terapéuticas.
The irruption of lipoprotein(a) (Lp(a)) in the study of cardiovascular risk factors is perhaps, together with the discovery and use of proprotein convertase subtilisin/kexin type 9 (iPCSK9) inhibitor drugs, the greatest novelty in the field for decades. Lp(a) concentration (especially very high levels) has an undeniable association with certain cardiovascular complications, such as atherosclerotic vascular disease (AVD) and aortic stenosis. However, there are several current limitations to both establishing epidemiological associations and specific pharmacological treatment. Firstly, the measurement of Lp(a) is highly dependent on the test used, mainly because of the characteristics of the molecule. Secondly, Lp(a) concentration is more than 80% genetically determined, so that, unlike other cardiovascular risk factors, it cannot be regulated by lifestyle changes. Finally, although there are many promising clinical trials with specific drugs to reduce Lp(a), currently only iPCSK9 (limited for use because of its cost) significantly reduces Lp(a).
However, and in line with other scientific societies, the SEA considers that, with the aim of increasing knowledge about the contribution of Lp(a) to cardiovascular risk, it is relevant to produce a document containing the current status of the subject, recommendations for the control of global cardiovascular risk in people with elevated Lp(a) and recommendations on the therapeutic approach to patients with elevated Lp(a).
Kare Berg fue el primero en identificar la presencia aumentada en el suero de un antígeno muy influenciado por la herencia, que denominó apolipoproteína a [apo(a)] y que localizó en las lipoproteínas de baja densidad (LDL) de pacientes con infarto de miocardio1–3. Diferentes estudios confirmaron que la apo(a) forma parte de una lipoproteína (Lp) que por lo demás se asemeja a las LDL. Curiosamente, la Lp(a) solo se ha identificado en erizos y en primates, incluyendo al ser humano4.
Estructura, síntesis y catabolismoLa estructura proteica de la Lp(a) contiene fundamentalmente apolipoproteína B100 (apoB) y apo(a), siendo precisamente esta combinación el rasgo distintivo de esta lipoproteína5. La clonación y la secuenciación del gen de apo(a) (LPA) en 1987 demostró que se trataba de una proteína hidrofílica y altamente glicosilada, con un elevado grado de homología con el gen del plasminógeno6. Contiene unos característicos dominios denominados kringles, que son estructuras de 80-90 aminoácidos organizados en un triple bucle. La apo(a) contiene 2 tipos de kringle (K), el IV y el V, así como un dominio proteasa mutado sin actividad proteolítica6. Mientras que en cada molécula de apo(a) existe solo una copia de KV y del dominio proteasa, existen 10 subtipos de kringle KIV, que se enumeran del 1 al 10 (KIV1-10). Todos ellos, menos el KIV-2, presentan una única copia por molécula. El KIV-2 puede en cambio presentar desde una copia a más de cuarenta, pero la variabilidad polimórfica puede darse también en un individuo al heredar lpa de ambos progenitores.
El número de kringles KIV-2 condiciona la longitud y el peso molecular total de la apo(a), que varía entre 300 y 800 KDa. No solo la longitud y el peso molecular de apo(a) depende del número de copias del KIV-2, sino que este es un importante determinante inverso de la concentración de Lp(a) en suero7. Así, a mayor número de copias de KIV-2 en apo(a), menor concentración sérica de Lp(a) en unidades molares.
Todavía no se conocen completamente los mecanismos de síntesis y catabolismo de la Lp(a). Se considera que la Lp(a) es básicamente de síntesis hepática. La interacción entre dominios ricos en lisinas entre apoB-100 y apo(a) precede a la formación de un único puente disulfuro entre ambas apolipoproteínas, que podría producirse a nivel extracelular8. El catabolismo de Lp(a) también parece fundamentalmente hepático, aunque tampoco se han identificado los receptores implicados. No parece que el receptor de LDL tenga un papel primordial en la retirada de la circulación de Lp(a)4. Algunos estudios han detectado pequeñas cantidades de apo(a) no unida a apoB en orina, aunque el papel del riñón en el catabolismo de Lp(a) tampoco está bien definido9.
Determinantes genéticos y ambientales de la concentración de Lp(a)Genéticos: número de repeticiones de KIV y SNPLa concentración de Lp(a) está determinada en torno a un 80% por la herencia codominante en el gen LPA4,10. Se ha calculado que entre un 19 y un 70% de la variabilidad de la concentración de Lp(a) depende del número de repeticiones de KIV-2. Así las isoformas cortas de apo(a) (de 10 a 22 copias de KIV-2) se asocian a 4-5 veces mayor concentración de Lp(a) que las isoformas largas (>22 copias de KIV-2)4,11. Ello es debido, probablemente, a una disminución de la eficacia de la síntesis de apo(a) en las isoformas largas más que a diferencias de catabolismo. Sin embargo, pueden existir diferencias de más de 200 veces en la concentración sérica de Lp(a) entre individuos no relacionados, con isoformas de similar longitud, así como diferencias de más de 2 veces en familiares con las mismas isoformas de apo(a)11. Por tanto, otras variantes genéticas, aparte del número de copias de KIV-2, también determinan la concentración de Lp(a)12. Así, se han descrito numerosos polimorfismos de un solo nucleótido (SNP) que aumentan o disminuyen la concentración de Lp(a), como los SNP rs10455872 y rs379822012. Como curiosidad, algunos de los SNP localizados en el gen LPA se heredan en desequilibrio de ligamiento con el número de copias de KIV-2.
Cabe también considerar que hasta un 70% de KIV-2 está codificado para zonas hipervariables poco accesibles a las tecnologías habituales de secuenciación13, por lo que este es un tema que requerirá de mayor estudio. Variantes genéticas en los loci APOE, CETP y APOH también pueden, en menor medida, modular la concentración sérica de Lp(a). Por ejemplo, el alelo APOE2 se asocia a niveles más bajos de Lp(a)4,11.
La etnia también es un determinante de la concentración sérica de Lp(a), siendo esta mayor, en orden creciente, en personas de etnia china, caucásica, sudasiática y negra. Estas diferencias parecen depender fundamentalmente, del número de repeticiones de KIV-24,11. La concentración de Lp(a) es bastante estable a lo largo de la vida adulta y ligeramente más alta (5-10%) en mujeres que en varones4,11.
Ambientales: fisiológicas (ayuno, dieta, ejercicio) y patológicas (alteración hepática, renal, inflamación y cambios hormonalesAunque no tan bien caracterizados como los genéticos, y en general de menor importancia, los factores ambientales también pueden modificar la concentración sérica de Lp(a), especialmente en algunas circunstancias fisiológicas y patológicas. Además, existen todavía factores asociados a la concentración de Lp(a) cuyo origen y significado clínico no están plenamente identificados. Como ejemplo, se ha identificado una relación inversa entre la concentración de Lp(a) y triglicéridos, especialmente cuando los triglicéridos son superiores a 400mg/dl14.
La concentración de Lp(a) no cambia sustancialmente con el ciclo ayuno-situación posprandial, ni con el ejercicio físico11. La dieta tampoco parece influir de forma notable la concentración de Lp(a), habiéndose descrito descensos de 10-15% en aquellas dietas pobres en hidratos de carbono y ricas en grasas. El embarazo duplica las concentraciones de Lp(a), mientras que hay un ligero aumento de Lp(a) en la menopausia11.
Como órgano de síntesis de Lp(a), las alteraciones hepáticas graves pueden alterar la concentración de esta11. Las alteraciones renales graves tienden a aumentar la Lp(a) a través del aumento de la síntesis hepática asociada a proteinuria o a un descenso del catabolismo renal15. Las enfermedades inflamatorias crónicas aumentan la concentración de Lp(a), mientras que, en situaciones clínicas graves, en las que hay riesgo vital y reacción de fase aguda, hay un descenso de la concentración de Lp(a)16,17.
Algunas hormonas que condicionan la concentración de lipoproteínas afectan también a la concentración de Lp(a)11. Así, las alteraciones tiroideas influencian la concentración de Lp(a) de la misma forma que al colesterol de LDL. Es decir, el hipertiroidismo disminuye su concentración, mientras que el hipotiroidismo la aumenta. La hormona del crecimiento y el tratamiento hormonal sustitutivo en la menopausia aumenta y disminuye, respectivamente, la concentración de Lp(a)11.
Para finalizar este apartado es importante recordar que, como hemos comentado anteriormente, los cambios por hormonas y reacción de fase aguda son relativamente modestos en comparación con los niveles definidos genéticamente.
Factores patogénicos (arteriosclerosis)Aunque la concentración sérica de Lp(a) es menor que la de LDL, hay indicios de que la patogenicidad de la primera podría ser mayor18. Esto podría explicarse en primer lugar por la tendencia al acúmulo de Lp(a) en la pared arterial, favorecido por la unión del dominio rico en lisinas de KIV-10 de apo(a) a las proteínas de la matriz extracelular19 y, en segundo lugar, por la capacidad de la Lp(a) tanto para iniciar la inflamación vascular, como para facilitar la progresión de la lesión arteriosclerosa20. Los fosfolípidos oxidados, que son mayoritariamente transportados en el suero por Lp(a), han sido propuestos como mediadores importantes en los mecanismos proinflamatorios y procalcificantes que induce la Lp(a) ya sea en la lesión arteriosclerosa de la pared vascular o a nivel de las válvulas cardíacas21.
La Lp(a) podría presentar también capacidad protrombótica por aumento de la coagulación o por inhibición de la fibrinólisis (p. ej., por competencia con el plasminógeno). Sin embargo, cambios drásticos de la concentración de Lp(a) inducidos por tratamientos no alteran la actividad fibrinolítica ex vivo22. En este sentido, no hay evidencia epidemiológica de asociación entre los niveles de Lp(a) elevados y mayor riesgo de trombosis venosa, un contexto diferente de las lesiones arteriosclerosas23.
Metodología de laboratorio para la determinación de Lp(a) en EspañaUno de los mayores retos para los próximos años en la presentación de la Lp(a) como un agente predictor de riesgo de enfermedad cardiovascular (ECV) y potencial diana terapéutica, es establecer una metodología estandarizada para su determinación en la práctica clínica. Esto permitiría realizar comparaciones entre distintas poblaciones, y establecer niveles de referencia y recomendaciones consistentes. Como veremos más adelante, esto aún no ha ocurrido, y existen varios métodos de determinación, e incluso diferentes unidades de medida no intercambiables (mg/dl, nmol/l) que dificultan en cierta medida la interpretación de los resultados de laboratorio y la posibilidad de realizar intervenciones terapéuticas. Su determinación en sí no requiere condiciones especiales, pudiendo realizarse en ayunas o no, aunque se recomienda no realizar su determinación durante procesos agudos a excepción del síndrome coronario agudo24.
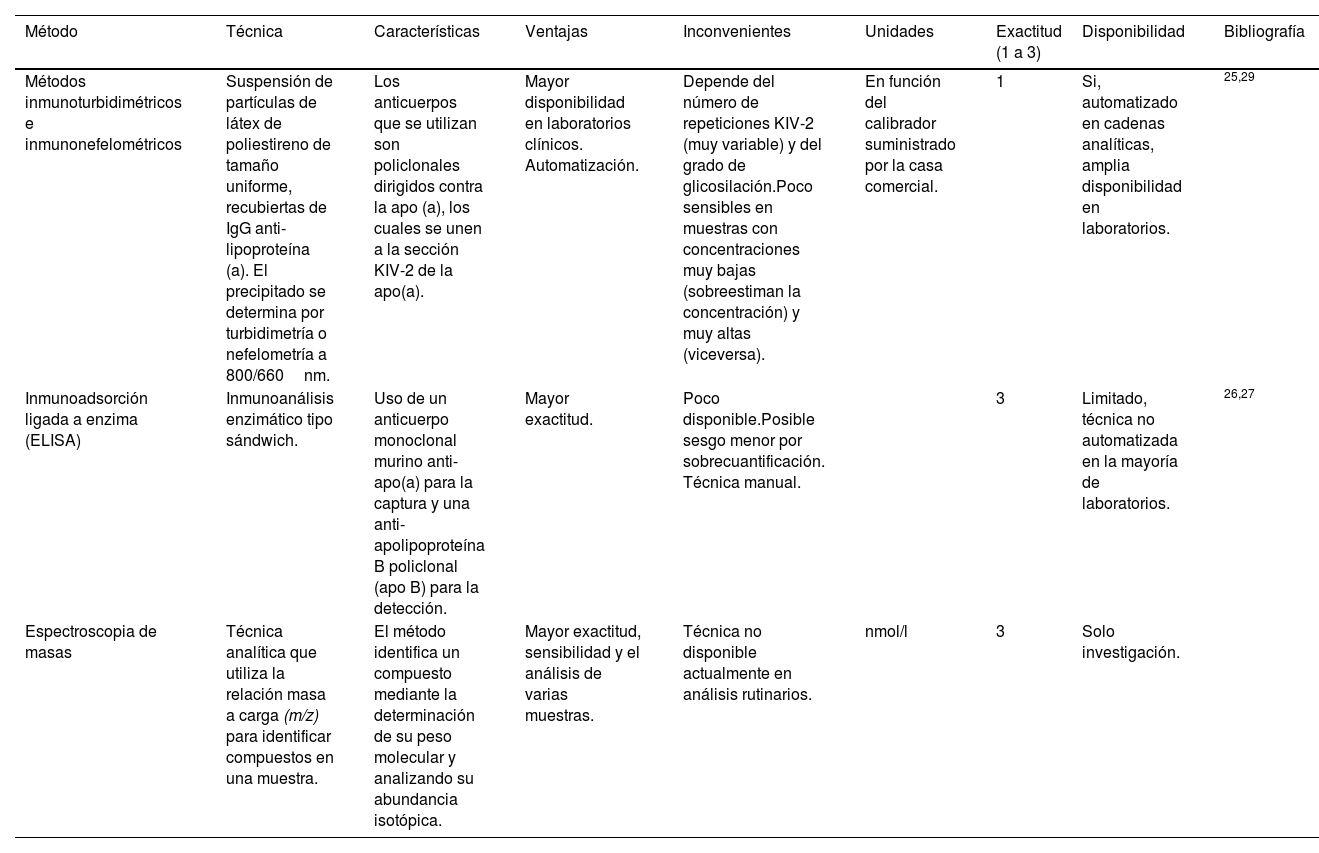
TécnicaExisten mayoritariamente en España 2 tipos de métodos de medición: los basados en la detección de KIV-2 y los basados en la medición de las moléculas ApoA y ApoB (explicados con mayor profundidad en la tabla 1).
Ensayos disponibles en España para la determinación de Lp(a)
| Método | Técnica | Características | Ventajas | Inconvenientes | Unidades | Exactitud (1 a 3) | Disponibilidad | Bibliografía |
|---|---|---|---|---|---|---|---|---|
| Métodos inmunoturbidimétricos e inmunonefelométricos | Suspensión de partículas de látex de poliestireno de tamaño uniforme, recubiertas de IgG anti-lipoproteína (a). El precipitado se determina por turbidimetría o nefelometría a 800/660nm. | Los anticuerpos que se utilizan son policlonales dirigidos contra la apo (a), los cuales se unen a la sección KIV-2 de la apo(a). | Mayor disponibilidad en laboratorios clínicos. Automatización. | Depende del número de repeticiones KIV-2 (muy variable) y del grado de glicosilación.Poco sensibles en muestras con concentraciones muy bajas (sobreestiman la concentración) y muy altas (viceversa). | En función del calibrador suministrado por la casa comercial. | 1 | Si, automatizado en cadenas analíticas, amplia disponibilidad en laboratorios. | 25,29 |
| Inmunoadsorción ligada a enzima (ELISA) | Inmunoanálisis enzimático tipo sándwich. | Uso de un anticuerpo monoclonal murino anti-apo(a) para la captura y una anti-apolipoproteína B policlonal (apo B) para la detección. | Mayor exactitud. | Poco disponible.Posible sesgo menor por sobrecuantificación. Técnica manual. | 3 | Limitado, técnica no automatizada en la mayoría de laboratorios. | 26,27 | |
| Espectroscopia de masas | Técnica analítica que utiliza la relación masa a carga (m/z) para identificar compuestos en una muestra. | El método identifica un compuesto mediante la determinación de su peso molecular y analizando su abundancia isotópica. | Mayor exactitud, sensibilidad y el análisis de varias muestras. | Técnica no disponible actualmente en análisis rutinarios. | nmol/l | 3 | Solo investigación. |
Lp(a): lipoproteína(a).
Los métodos inmunoturbidimétricos e inmunonefelométricos son los más utilizados en nuestro medio. La principal característica de los que están al alcance de la mayoría de los centros es que emplean inmunoglobulinas que se unen a la sección KIV-2 de la apo(a). Como ya ha sido comentado, KIV-2 es una secuencia que puede estar representada en mayor o menor número de repeticiones dentro de la partícula de apo(a) (véase el apartado «Definición, estructura y regulación»), lo que puede limitar parcialmente la precisión de la prueba. Estos métodos están evolucionando para poder reaccionar con anticuerpos contra una estructura con representación única en la molécula de Lp(a), como el KV o el dominio de proteasa25.
Menos disponible, existe el ensayo inmunoadsorbente ligado a enzimas (ELISA), que es teóricamente es más preciso. Tras el atrapamiento de Lp(a) con un anticuerpo frente a la apo(a), la cuantificación se realiza mediante un anticuerpo frente a apo B, dado que existe una única molécula de cada una de ellas en la partícula de Lp(a). De este modo se anula la imprecisión debida a la medición de secuencias variables (que determinan las diferentes isoformas) dentro de la molécula de apo(a)26 (tabla 1). Algunos autores señalan la posible sobreestimación de Lp(a) en ensayos de este tipo, basados en la captura de apo(a)/detección de apoB, debido a la unión marginal que pueden tener las partículas de Lp(a) asociadas de forma no covalente a lipoproteínas ricas en triglicéridos27.
En relación con los métodos existentes, y con referencia a los basados en la detección de KIV-2, cabe recordar que la concentración de Lp(a) está fuertemente determinada genéticamente, con una influencia mínima de la dieta o de la modificación del estilo de vida. Parte de la influencia genética sobre la Lp(a) de la descendencia es el número de repeticiones de KIV-2 (véase la sección sobre genética de la Lp(a)). Las partículas con un mayor número de repeticiones KIV-2 son más grandes y pesadas (se ha referido que podría encontrarse una diferencia estimada de masa del 19% entre una partícula de Lp(a) con 6 repeticiones KIV-2 en apo(a) comparada con la Lp(a) con 35 repeticiones KIV-2, por lo que la medición de la concentración de Lp(a) puede estar artefactada cuando se utilizan ensayos que no son independientes de la isoforma28. Estos errores de medición afectan en mayor medida a las cuantificaciones con resultados extremos, infraestimando las concentraciones de muestras procedentes de personas con isoformas de Lp(a) pequeñas (asociadas a niveles más elevados de Lp(a)) y viceversa29. Esto es especialmente importante porque de este modo se estaría infraestimando el riesgo de pacientes con isoformas de la Lp(a) pequeñas y viceversa11.
Unidades y conversión de medidasLa mayoría de los consensos, enfoques terapéuticos y guías de práctica clínica actuales expresan las mediciones de Lp(a) en unidades de masa (mg/dl). Esto se debe principalmente a razones didácticas e históricas, ya que los profesionales clínicos están más acostumbrados a su uso. Para ello, a menudo se utilizan sistemas de conversión a partir de las unidades originales expresadas por las diferentes pruebas analíticas mencionadas anteriormente. Sin embargo, en este caso, este enfoque no es realmente correcto desde el punto de vista metrológico. La razón es que estas pruebas analíticas solo determinan la concentración de la fracción proteica de la apo(a), y no la masa de los demás componentes de la partícula de Lp(a), como el colesterol, los ésteres de colesterol, los fosfolípidos, los triglicéridos y los hidratos de carbono. Además, al convertir nmol/l a mg/dl, se comete un segundo error metrológico, ya que las isoformas de apo(a) tienen un peso molecular variable, lo que no permite una conversión unívoca entre masa y unidades molares.
A efectos prácticos, en el resto del documento se seguirán utilizando las unidades habitualmente empleadas en la literatura (mg/dl), aunque el lector de este documento debe ser consciente de que este enfoque produce cierto margen de error, especialmente en los valores extremos de Lp(a). Es probable que esto mejore con la introducción de test más precisos, sensibles a las isoformas y basados en unidades molares de apo(a) que reflejen la cantidad de partículas de Lp(a) presentes en el suero/plasma de un individuo, independientemente de la cantidad de repeticiones de KIV-2, el grado de N - y O-glicosilación de apo(a) o la composición lipídica de Lp(a). Esto permitirá establecer límites de normalidad y puntos de corte más precisos para establecer las distintas acciones terapéuticas que se expresen en las unidades correctas (nmol/l)29–33.
Varias iniciativas en curso, en esta dirección, tratan de resolver los problemas metrológicos en la medida más exacta de la Lp(a) mediante la fabricación de materiales calibrados en unidades molares trazables a las referencias verificadas (OMS/IFCC, Northwest Lipid Metabolism and Diabetes Research Laboratory (NLMDRL), Federación Internacional de Química Clínica y Medicina de Laboratorio (IFCC)34.
Para concluir, se han presentado recientemente varios trabajos que avanzan en la protocolización de la determinación de Lp(a) por espectrometría de masas con los más avanzados estándares de calidad35,36.
Hasta que estos test sean una realidad, la Sociedad Europea de Arteriosclerosis (EAS) recomienda que se incluyan los valores de referencia específicos del test realizado en los informes de laboratorio. Aunque como se ha indicado anteriormente, no se recomienda realizar conversión entre unidades, reconocemos que en determinadas ocasiones los clínicos necesitan tener una orientación del resultado de la prueba de su paciente con respecto a las guías de práctica clínica y consensos. En este sentido, los factores de conversión de mg/dl a nmol/l suelen oscilar entre 1:2,0 y 1:2,511,37, aunque, como se ha comentado, esta conversión es poco exacta y solo debe realizarse para una aproximación burda al resultado de la prueba y probablemente solo para determinar si un paciente está en una zona muy alta o muy baja38. El lector del presente documento debe de tener en cuenta lo anterior, por lo que en distintas partes de este documento pueden aparecer distintos factores de conversión. Esto viene derivado de que se han intentado respetar las conversiones realizadas en los artículos originales.
En resumen, y dada la heterogeneidad de la determinación de Lp(a) en el momento actual, deberían remarcarse algunos conceptos clave, como que las concentraciones séricas de Lp(a) deben medirse idealmente utilizando un método donde el efecto del tamaño de la isoforma se haya minimizado utilizando anticuerpos apropiados con calibradores certificados para la trazabilidad de valores de Lp(a) al material de referencia de la OMS/IFCC, y que si el laboratorio de trabajo del facultativo no posee este tipo de métodos, se debe conocer que la exactitud de la prueba es inferior. También, que los resultados deben expresarse idealmente en nmol/L de partículas de Lp(a), y que si se utilizan otras unidades siempre deben aparecer los límites de referencia del test utilizado. Finalmente, hay que recalcar que la conversión de unidades de masa a unidades molares o viceversa, introduce inexactitud, y que siempre que se utilice debe de conocerse que los resultados obtenidos solo son orientativos.
Lipoproteína(a) y su relación con la enfermedad vascular, la estenosis aórtica, y otras enfermedadesDesde el descubrimiento de la Lp(a) se han publicado numerosos estudios epidemiológicos que la relacionan con el riesgo de enfermedad vascular aterosclerótica (EVA)1. Los primeros estudios prospectivos, en la década de los noventa del siglo pasado, no encontraron una correlación consistente con la EVA o el infarto agudo de miocardio (IAM), probablemente debido al uso de métodos de medición poco precisos39. Esto llevó a una pérdida de interés científico por la Lp(a) en los años posteriores, con una importante disminución de las publicaciones sobre la misma en ese periodo. Sin embargo, los estudios epidemiológicos y genéticos más recientes sugieren una asociación causal entre los niveles elevados de Lp(a), la EVA, la estenosis aórtica (EA), así como otras patologías. La mayoría de la población tiene concentraciones de Lp(a) bajas o muy bajas, pero se estima que entre el 20-25% presenta niveles mayores de 50mg/dl, un valor que la Sociedad Europea de Aterosclerosis considera que confiere un mayor riesgo de morbimortalidad ECV11.
Estudios epidemiológicos que relacionan la Lp(a) con la enfermedad coronariaEn la primera década del presente siglo se publicaron grandes estudios prospectivos de población general. El Copenhagen City Heart Study siguió a 9.339 individuos daneses durante 10 años, y observó que concentraciones crecientes de Lp(a) se asociaban de forma independiente con el riesgo de enfermedad arterial coronaria (EAC), aumentando en un 9% el riesgo de IAM por cada incremento de 10mg/dl en la concentración de Lp(a)40; así, en comparación con una concentración de Lp(a)<5mg/dl, un nivel de Lp(a)≥120mg/dl confería un riesgo relativo de infarto casi 4 veces mayor. En este mismo estudio se demostró que una concentración elevada de Lp(a) se asociaba a una mayor mortalidad CV y por cualquier causa, independientemente de la concentración de colesterol de lipoproteínas de baja densidad (c-LDL)41. En 2009 un metaanálisis de 36 cohortes prospectivas con 126.634 participantes sin EVA previa, elaborado por el Emerging Risk Factors Collaboration, confirmó una asociación continua e independiente, aunque moderada, de la concentración de Lp(a) con el riesgo de IAM no fatal y con la muerte CV42. Aunque este metaanálisis incluyó principalmente estudios realizados en población caucásica, también hubo representación de otros grupos raciales, no encontrándose diferencias en las estimaciones de riesgo de los diferentes grupos étnicos. En el posterior estudio, Atherosclerosis Risk in Communities (ARIC), realizado en población blanca y negra, los niveles de Lp(a) se asociaron positivamente con la EVA con la misma fuerza en ambos grupos raciales, aunque la mediana de concentraciones de Lp(a) fue 2-3 veces más alta en personas de raza negra con relación a las personas de raza blanca43. El análisis de 460.506 individuos de mediana edad de la base de datos del UK Biobank en seguimiento durante una mediana de 11,2 años, mostró una asociación lineal entre la concentración de Lp(a) y el riesgo de EVA (hazard ratio [HR]: 1,11; IC del 95%: 1,10-1,12) por cada 50nmol/l de aumento44.
También en individuos con hipercolesterolemia familiar (HF) se ha observado un mayor aumento de riesgo de EAC en aquellos que presentan concentraciones de Lp(a) superiores a 50mg/dl cuando se los comparaba con individuos con HF y niveles de Lp(a) menores de esta cifra45.
Los niveles de Lp(a) elevados se han relacionado también con la recurrencia de episodios CVs en individuos con EAC establecida, si bien los datos en estas poblaciones han sido inconsistentes46. En sujetos daneses en prevención secundaria, la concentración creciente de Lp(a) se asoció con un mayor riesgo de recurrencias, estimándose que sería necesario reducir en 50mg/dl (105nmol/l) su concentración durante 5 años, para obtener una reducción de la tasa de eventos CVs del 20%47. En un metaanálisis de 18.979 individuos, los niveles de Lp(a)>percentil 80 fueron predictores de episodios CVs recurrentes en pacientes con EAC tratados con estatinas y se asociaron con un 40% más de eventos CVs mayores (MACE); pero solo cuando el c-LDL inicial era ≥130mg/dl48.
En definitiva, los estudios epidemiológicos demuestran que una concentración aumentada de Lp(a) se asocia con un mayor riesgo de EVA, fundamentalmente EAC, siendo mayor la magnitud en poblaciones en prevención primaria. La asociación entre variantes genéticas asociadas a Lp(a) elevada y eventos CVs desaparece cuando se ajustan por los niveles de Lp(a), sugiriendo un valor patogénico directo de Lp(a) en el desarrollo de la EAC49.
Estudios epidemiológicos que relacionan a la Lp(a) con la estenosis aórtica (EA)La EA calcificada es la forma más común de enfermedad cardíaca valvular adquirida que requiere intervención. Se estima que la prevalencia de EA es del 2 al 4% en individuos mayores de 65 años y se prevé que el número de personas con indicación de reemplazo de válvula aórtica se duplique en 205050. En los últimos años, el conocimiento de los mecanismos fisiopatológicos que llevan al desarrollo de esta enfermedad ha evolucionado desde una calcificación degenerativa, a un proceso activo derivado de la interacción de factores genéticos y la inflamación crónica, favorecida por factores de riesgo como el tabaquismo, la hipertensión arterial y la hipercolesterolemia. Aunque existe una superposición de factores de riesgo entre la EA calcificada y la EAC, ≈50% de los pacientes con EA calcificada no tienen EAC concomitante. Además, las estatinas, que tienen un efecto beneficioso significativo sobre la EVA, hasta el momento no han demostrado detener la progresión de la EA51,52, lo que sugiere la existencia de otros factores fisiopatológicos en la EA calcificada.
Un trabajo publicado en 2013 fue el primero que estableció que los niveles de Lp(a) derivados de variaciones genéticas en el locusLPA se asociaban a la calcificación de la válvula aórtica y a la EA clínica53. Un estudio posterior en 77.680 participantes, que combinaba datos del Copenhagen City Heart Study y el Copenhagen General Population Study, mostró que los niveles elevados de Lp(a) estaban asociados con un mayor riesgo de EA, y que niveles de Lp(a)>90mg/dl multiplicaban por 3 el riesgo de padecerla54. Dichos hallazgos han sido corroborados en posteriores estudios epidemiológicos. Más recientemente, una revisión sistemática de 21 estudios, incluyendo estudios de casos y controles, observacionales prospectivos y retrospectivos, y genéticos, ha demostrado una asociación significativa entre Lp(a) elevada y EA calcificada; además, la Lp(a) elevada predice una progresión hemodinámica acelerada de la EA y un mayor riesgo de reemplazo valvular aórtico, especialmente en pacientes jóvenes55. El aumento de la Lp(a) se asocia con la calcificación de la válvula aórtica sobre todo en individuos jóvenes (45-54 años), en los que el riesgo se triplica cuando los niveles de Lp(a) están por encima del percentil 80 frente a niveles inferiores (15,8% frente a 4,3%)56. En un subestudio del ensayo FOURIER, que incluyó a 27.564 pacientes con EVA que tomaban estatinas, los niveles elevados de Lp(a), pero no los niveles de c-LDL, se asociaron con un mayor riesgo de eventos posteriores de EA, ya sea progresión o necesidad de cirugía aórtica con reemplazo valvular57.
Por tanto, la elevación de la concentración de Lp(a) se asocia con el desarrollo de EA, sobre todo en personas jóvenes, con una mayor velocidad de progresión y con un mayor riesgo de sustitución valvular.
Estudios epidemiológicos que relacionan a la Lp(a) con la insuficiencia cardíaca (IC) y la fibrilación auricularEl análisis combinado del Copenhagen General Population Study y el Copenhagen City Heart Study, demostró que niveles de Lp(a) por encima del percentil 75 y ciertos genotipos de riesgo del gen LPA (como rs3798220 y rs10455872) aumentaban el riesgo de IAM y EA, y que los niveles superiores al percentil 90 se asociaban con un mayor riesgo de IC, la cual parecía estar parcialmente mediada por el mayor riesgo de IAM y EA58.
La asociación entre la EVA y fibrilación auricular es bien conocida. Sin embargo, se desconoce si la Lp(a) es un factor causal independiente de fibrilación auricular. En un estudio procedente de la cohorte del UK Biobank, y que reclutó a más de 500.000 personas de entre 37 y 73 años en todo el Reino Unido entre 2006 y 2010, los autores concluyeron que se podía establecer una potencial implicación de la Lp(a) en el riesgo de desarrollar fibrilación auricular, que era parcialmente independiente del efecto ateroesclerótico59.
Estudios epidemiológicos que relacionan a la Lp(a) con la arteriosclerosis en otros territoriosLa relación entre la Lp(a), el ictus y la enfermedad arterial periférica (EAP) es plausible desde el punto de vista fisiopatológico, ya que la aterotrombosis constituye la principal base etiopatogénica de ambas patologías, de forma similar a lo que ocurre en la enfermedad coronaria. Así, el efecto aterogénico de la Lp(a) se ejercería en los distintos territorios arteriales, ya a través de la LDL que forma parte de la molécula de Lp(a), el efecto proinflamatorio de los fosfolípidos oxidados que contiene, o la acción antifribrinolítica y trombogénica de la apo(a)4.
La relación entre el ictus y los factores de riesgo de ECV es más compleja que en el caso de la EAC o la EAP. Ello obedece a que el ictus puede tener distintas etiologías, incluyendo, además de la aterotrombosis, la hemorragia, el embolismo o la hialinosis arteriolar. A pesar de ello, existe una amplia base de datos derivada de los estudios epidemiológicos que demuestra que un aumento de la concentración de Lp(a) se asocia a un mayor riesgo de ictus isquémico. Estudios a gran escala demuestran que existe una relación entre la concentración de Lp(a) y el riesgo de ictus. En el Copenhagen General Population Study (n=49.699) y el Copenhagen City Heart Study (n=10.813)60 las concentraciones séricas de Lp(a) se relacionaron gradual, directa y continuamente con el riesgo de ictus. Esta relación ha sido confirmada en grandes metaanálisis, entre ellos el Emerging Risk Factors Collaboration, con un seguimiento de 1,3 millones de personas/año, en el que se observó una relación continua entre la concentración de Lp(a) y el riesgo de ictus42. En dicho metaanálisis, un incremento de 3,5 veces en la concentración de Lp(a) (1 desviación estándar) se asoció a un riesgo de ictus isquémico de 1,10 (IC 95%: 1,02-1,18), que fue independiente de otros factores de riesgo de ECV. Así mismo, en un metaanálisis de 20 estudios (n=90.904 individuos), las concentraciones elevadas de Lp(a) se asociaron a una odds ratio (OR) de ictus isquémico de 1,41 (IC 95%: 1,26-1,57) en los estudios de casos y controles y de 1,29 (IC 95%: 1,06-1,58) en los estudios prospectivos61. En coherencia con estos datos, las concentraciones de Lp(a) también se relacionan con el riesgo residual de recurrencia del ictus en los pacientes que sufren un primer episodio de ictus isquémico62, si bien la relación podría atenuarse en los pacientes con unas concentraciones bajas de c-LDL o de marcadores de inflamación63.
La relación entre Lp(a) y EAP ha sido demostrada en estudios prospectivos de amplios grupos de población, entre ellos el Edinburgh Artery Study64, el Scottish Heart Health Extended Cohort Study65 y el estudio EPIC66. Este último fue realizado en una amplia cohorte de individuos sanos (n=18.720) y en él, un incremento de 2,7 veces en la concentración de Lp(a) se asoció a un aumento del riesgo de esta enfermedad de 1,37 (IC 95%: 1,25-1,50), independientemente de los valores de c-LDL.
En un análisis del registro español FRENA entre 1.503 pacientes ambulatorios estables con EAC, cerebrovascular o EAP, los que presentaban unas concentraciones de Lp(a) entre 30 y 50mg/dl sufrieron una mayor tasa de amputaciones de extremidades que los que presentaban unas concentraciones inferiores a 30mg/dl (riesgo relativo [RR]: 3,18; IC 95%: 1,36-7,44). El riesgo de amputación fue aún mayor cuando la concentración de Lp(a) subía de 50mg/dl (RR: 22,7; IC 95%: 9,38-54,9)67. Así mismo, en una revisión sistemática que incluía 493.650 sujetos, se observó que una concentración elevada de Lp(a) se asociaba a un mayor riesgo de claudicación intermitente (RR: 1,20), progresión de la EAP (RR: 1,41), reestenosis (RR: 6,10), muerte y hospitalización relacionada con la EAP (RR: 1,37), amputación (RR: 22,75) y revascularización de miembros inferiores (RR: entre 1,29 y 2.90 en función de factores como el punto de corte de Lp(a) y los años de seguimiento), independiente de los factores de riesgo de ECV tradicionales68.
Cuanto mayor es la concentración de Lp(a), mayor es el riesgo de presentar manifestaciones clínicas de arteriosclerosis en más de un territorio arterial, es decir una arteriosclerosis generalizada, y también mayor es la gravedad de las manifestaciones clínicas de la enfermedad69, incluyendo una mayor mortalidad por todas las causas70. Los pacientes con un síndrome coronario agudo que son sometidos a una intervención coronaria percutánea que presentan una EAP, tienen unas concentraciones de Lp(a) más elevadas que los que no la presentan71. En el mismo sentido, los pacientes intervenidos de EAP o de cirugía carotídea que presentan una Lp(a)>30mg/dl tienen un riesgo de ictus, IAM o muerte de causa ECV 3 veces superior (IC 95%: 1,5-6,3) a los que tienen unas concentraciones de Lp(a) dentro del intervalo de referencia70. El aumento de Lp(a) también se asocia a un mayor riesgo de reestenosis con necesidad de reintervención o de amputación después de las intervenciones de revascularización arterial de las extremidades inferiores72. En estos pacientes, una concentración elevada de Lp(a) comporta un mayor riesgo de ictus, EAC o muerte de causa ECV. Por último, algunos estudios de casos y controles sugieren que las concentraciones elevadas de Lp(a) se relacionan con la disección aórtica73.
En definitiva, la concentración elevada de Lp(a) se asocia con un mayor riesgo de ictus aterotrombótico y de EAP, así como una mayor velocidad de progresión y riesgo de amputación en los pacientes con EAP.
Estudios epidemiológicos que relacionan a la Lp(a) con la diabetesEstudios realizados en distintas poblaciones han sugerido que existe una relación inversa entre las concentraciones de Lp(a) y el riesgo de desarrollar diabetes mellitus tipo 2. Sin embargo, esta relación no es lineal en toda la distribución de valores de Lp(a), sino que se manifiesta de forma acusada con concentraciones muy bajas de Lp(a) y deja de ser evidente con concentraciones medias o altas74. En un estudio de casos y controles realizado en 143.087 individuos de la población islandesa, el 10% de los que tenían las concentraciones más bajas de Lp(a) (<3,5nmol/l) tenía una mayor prevalencia de diabetes mellitus tipo 2 (OR: 1,44; p<0,0001). Sin embargo, en los que tenían unas concentraciones superiores a la mediana, el riesgo de diabetes no se asociaba con las concentraciones de Lp(a)75. En otro estudio prospectivo de población general, también se observó que unas concentraciones de Lp(a) en el quintil inferior (<10nmol/l) se asociaban a un mayor riesgo de diabetes y el riesgo se incrementaba aún más en los que tenían concentraciones de Lp(a) todavía más bajas76. En otro metaanálisis de 201777, se mostró que los sujetos con valores bajos de Lp(a), entre 3 y 5mg/dl, tenían un riesgo de diabetes un 38% superior al de los sujetos con valores de entre 27 y 55mg/dl.
Aparentemente, solo una parte de la asociación de la Lp(a) con la diabetes se debe a una relación de causalidad, sin que se haya podido definir si la relación se debe a las concentraciones de Lp(a), a la longitud de las isoformas de la apo(a) o a una combinación o interacción entre ambas. La relevancia de descifrar si existe una relación de causalidad entre las concentraciones muy bajas de Lp(a) y el riesgo de diabetes se debe a que, si existiera una evidencia firme de la misma, cabría plantearse si disminuir la Lp(a) a concentraciones muy bajas con los nuevos fármacos en desarrollo podría aumentar el riesgo de diabetes. Con todo, los datos de los estudios epidemiológicos sugieren que para aumentar el riesgo de diabetes deberían disminuirse las concentraciones de Lp(a) a valores extremadamente bajos, lo que parece poco probable74. Por otro lado, ha sido bien demostrado que el exceso de Lp(a) predispone a las complicaciones micro y macrovasculares en la población diabética y los análisis de los grandes ensayos clínicos con iPCSK978 sugieren que en la población diabética o prediabética el potencial riesgo diabetogénico del descenso de la Lp(a) sería ampliamente superado por el beneficio ECV del marcado descenso del colesterol aterogénico79.
Contribución genética de la Lp(a) a la tasa de complicaciones CVsComo hemos visto anteriormente, entre un 80-90% de la concentración de Lp(a) es determinada genéticamente. Esto permite relacionar ciertas variantes genéticas en el gen LPA, responsables mayoritarias de la concentración de Lp(a) en plasma, con la tasa de complicaciones CVs sin que esta relación se encuentre artefactada por factores externos, lo que indicaría una relación de causalidad80. Además de las isoformas asociadas con variantes genéticas que regulan el número de repeticiones del KIV-2, otras variantes que se han relacionado con incrementos en la concentración de Lp(a) son rs10455872 y rs3798220.
Contribución genética de la Lp(a) a la EACDiversos estudios de asociación genética y aleatorización mendeliana, han demostrado una relación entre diferentes variantes en LPA con la gravedad y extensión de la arteriosclerosis81, y con el riesgo de desarrollar complicaciones vasculares, principalmente coronarias. Muchos de estos estudios se han realizado en la población danesa, habiéndose observado que el número de repeticiones del KIV-2 se asocia de forma inversa con el riesgo de IAM82. Sin embargo, si bien la asociación con la mortalidad CV y total se observó con el número de repeticiones, esta asociación no se observó con la variante rs1045587241. En este mismo estudio se calculó la cantidad de colesterol de las partículas de Lp(a). Para una misma cantidad de colesterol, la magnitud de la asociación de la Lp(a) con la mortalidad total y CV fue mayor que la observada para las LDL, lo que implica que el efecto de la Lp(a) sobre la mortalidad es mayor del atribuible a su contenido de colesterol.
Estudios realizados en otras poblaciones y grandes metaanálisis han corroborado dichas asociaciones. En un metaanálisis de 40 estudios, la presencia de isoformas pequeñas de apo(a) duplicó la tasa de infarto y de ictus en comparación con la presencia de isoformas mayores de 22 repeticiones del KIV-283. En otros estudios donde se han evaluado variantes genéticas asociadas con Lp(a) aumentada, se demostró que la elevación de Lp(a) genéticamente predicha se asociaba con EAC49,82. Variantes genéticas asociadas con elevación de la Lp(a) también predicen el riesgo de EAC en sujetos ya tratados con estatinas, independientemente del descenso de c-LDL inducido por estas84.
Otros estudios han demostrado que variantes alélicas asociadas con pequeñas reducciones de concentración de Lp(a), protegen del desarrollo de EAC85,86. De hecho, se ha podido demostrar que por cada diferencia de 10mg/dl en la concentración de Lp(a) determinada genéticamente, se reduce la tasa de EAC en un 5,8%.
Se han elaborado scores genéticos con diversas variantes de Lp(a) para mejorar la predicción del riesgo de complicaciones CV87. Si bien su predicción es adecuada, la fuerza de la asociación se reduce significativamente al incluir en la ecuación la concentración de Lp(a), con lo que su utilidad podría ser limitada, a lo que se añade la especial dificultad de realizar estudios genéticos de las zonas hipervariables de los KIV12.
En prevención secundaria no se ha demostrado que en los sujetos con EAC, las variantes genéticas de LPA tengan un impacto sobre la mortalidad88.
Contribución genética de la Lp(a) a la estenosis de válvula aórticaEn el estudio EPIC, la variante rs10455872 se asoció con una mayor incidencia de EA89, un dato corroborado en un metaanálisis que incluyó los datos del UK Biobank90. En este trabajo, el riesgo de EA aumentó entre un 10 y un 30% por cada incremento en 10mg/dl de la concentración de Lp(a) determinada genéticamente. Por el contrario, la variante rs3798220, con una menor frecuencia poblacional, se ha asociado de forma irregular con el riesgo de EA90. En un gran estudio de casos y controles que incluyó a 3.469 sujetos con EA, ambas variantes se asociaron con un mayor riesgo de padecerla91. La relación de ambos polimorfismos ha sido también estudiada en un reciente metaanálisis92 que demostró que los sujetos con estas variantes genéticas tenían progresiones más rápidas y mayor riesgo de complicaciones clínicas, incluida la muerte.
Contribución genética de la Lp(a) a otras patologías vascularesEn el Multiancestry Genome-Wide Association Study of Stroke consortium (n ≤446.696), en el que se analizó la relación de 9 polimorfismos asociados con las concentraciones de Lp(a) con el riesgo de distintos subtipos de ictus, las variantes que cursaban con un incremento de las concentraciones de Lp(a) se asociaban a un mayor riesgo de ictus de grandes arterias93. Por cada aumento de una desviación estándar en las concentraciones de Lp(a), la OR de ictus de grandes arterias era de 1,20 (IC 95%: 1,11-1,30; p<0,001). Sin embargo, el riesgo de ictus de pequeño vaso era menor (OR: 0,92; IC 95%: 0,88-0,97); p=0,001) y no hubo asociación con el conjunto de ictus isquémicos o de origen cardioembólico. En este estudio también se observó una relación inversa entre las concentraciones de Lp(a) genéticamente determinadas y la incidencia de enfermedad de Alzheimer, un aspecto sobre el que existen datos controvertidos y pueden intervenir distintos factores de confusión93. La relación entre variantes genéticas y riesgo de ictus también se ha demostrado en la población danesa, donde el número de repeticiones del KIV y la variante rs10455872, se asocian con un incremento del riesgo de ictus, del 20 y 27%, respectivamente60.
La elevación genética de la Lp(a) también se ha asociado con el riesgo de desarrollar insuficiencia cardiaca58, un efecto parcialmente mediado por el mayor riesgo de EAC y de EA.
El gen LPA se ha asociado también con el desarrollo de EAP en estudios de aleatorización mendeliana y de asociación del genoma completo23,94–96. En referencia a estos últimos, en el Million Veteran Program se analizó la asociación de 32 millones de variantes de las secuencias de ácido desoxirribonucleico (ADN) con la EAP (31.307 casos y 211.753 controles) en individuos de ascendencia europea, africana e hispana95. Los resultados se replicaron en una muestra independiente de 5.117 casos de EAP y 389.291 controles del UK Biobank. Se identificaron 19 loci asociados a la EAP, entre los que se encontraban 18 no identificados previamente. Entre ellos se incluían loci del gen LPA, lo que apoya que la Lp(a) se relaciona con la arteriosclerosis de las extremidades inferiores.
Por tanto, la Lp(a) ha de ser considerada como un biomarcador de utilidad en la valoración del riesgo de ictus aterotrombótico y de EAP. Con los nuevos fármacos actualmente en desarrollo, la Lp(a) será probablemente una diana terapéutica en la prevención primaria y secundaria de dichas patologías.
Contribución genética de la Lp(a) a otras enfermedadesSe ha descrito un menor riesgo de diabetes97 con concentraciones elevadas de Lp(a), que parece depender fundamentalmente de la variante genética, en concreto del número de repeticiones del KIV-2.
Por último, existe controversia sobre la relación entre Lp(a) y el riesgo de enfermedad tromboembólica venosa. Si bien el número de repeticiones del KIV tipo 2 se ha asociado de forma inversa con dicho riesgo98, estudios de aleatorización mendeliana no parecen confirmar dicha relación80.
Mediciones de Lp(a): cuándo y a quiénLa determinación de los niveles de Lp(a) tiene actualmente un doble interés: 1) la mejora de la estimación del riesgo CV en conjunto con otros factores de riesgo; 2) la evaluación de un factor de riesgo emergente, bastante estable en el tiempo, que permita tener identificada a la población, de cara a potenciales futuras aproximaciones terapéuticas. La ausencia de tratamientos efectivos para reducir la concentración de Lp(a) de forma relevante (en particular, la ausencia de estudios de intervención con variable final de resultados clínicos relevantes, específicamente eventos CV) y las dificultades en la estandarización de las mediciones de Lp(a) han retrasado la incorporación sistemática de su determinación en las guías de prevención de la ECV11,99–104.
El interés renovado en la Lp(a) como factor de riesgo vascular independiente y las expectativas de reducción farmacológica de sus niveles derivadas de ensayos clínicos en fases tempranas, han estimulado la progresiva incorporación de la propuesta de medición de los niveles de Lp(a) en determinados pacientes. Las recomendaciones iniciales fueron enfocadas a circunstancias concretas: ECV precoz (personal o familiar), en especial en ausencia de otros factores de riesgo CV, historia familiar de elevación de Lp(a), hipercolesterolemia familiar y pobre respuesta hipolipemiante a estatinas11,99–104. Dada la mayor accesibilidad de la determinación de Lp(a) en los laboratorios convencionales y el reconocimiento creciente de su valor pronóstico a partir de estudios observacionales, la tendencia actual, iniciada por las guías canadienses, es incluir la medición de Lp(a) como parte de la primera evaluación del riesgo CV global de todos los pacientes11,102,105. Por tanto, su determinación se incluye como parte de las recomendaciones de evaluación CV global inicial, teniendo que ser valorada en el contexto de la edad, sexo, presencia de otros factores de riesgo CV e historia familiar de ECV precoz del paciente. Dado que los niveles de Lp(a) están condicionados genéticamente y en general se mantienen estables a lo largo de la vida, como norma general se considera suficiente realizar una única determinación para la toma de decisiones clínicas11,106. No obstante, es razonable plantear una determinación de «confirmación» en determinadas circunstancias: Niveles muy elevados, dudas sobre la fiabilidad del método empleado, coexistencia con situaciones que pueden alterar su concentración, como el desarrollo de insuficiencia renal avanzada o síndrome nefrótico, si la primera determinación se realizó en la infancia, embarazo, disfunción hepatocelular/tiroidea importante, sepsis o proceso inflamatorio agudo, tal y como se expone en el material adicional, tabla S1.
Esta es la aproximación aceptada en el reciente consenso sobre el perfil lipídico básico coliderado por la Sociedad Española de Arteriosclerosis (SEA), la Sociedad Española de Medicina de Laboratorio y la Sociedad Española de Cardiología99 en colaboración con 15 sociedades científicas.
En pacientes en prevención secundaria en los que no se conozcan los niveles de Lp(a), es necesaria su determinación, en primer lugar, por su valor pronóstico, que puede aconsejar un reforzamiento de medidas preventiva o terapéuticas, como el uso de iPCSK9107. Adicionalmente, puede servir para identificar a pacientes candidatos a ensayos clínicos o tratamientos emergentes dirigidos específicamente a su reducción.
Como se ha mencionado con anterioridad, los niveles de Lp(a) están determinados genéticamente en un 80%. Puede plantearse si el genotipo de Lp(a) ofrece valor pronóstico superior al de la medición directa de la Lp(a) circulante. Aunque algunos polimorfismos genéticos (SNP rs10455872 y rs3798220) son relativamente frecuentes y se asocian a Lp(a) ‘pequeñas’ y niveles circulantes más elevados, más del 70% de la variabilidad (>500 variantes) está codificada en las regiones hipervariables de las repeticiones de los KIV-2, y su determinación es de especial dificultad para las tecnologías de secuenciación convencionales11,12. Adicionalmente, otros genes (lociAPOE, CETP, APOH) se asocian con cambios en la concentración de Lp(a). Finalmente, no se ha demostrado que el estudio del genotipo aporte información pronóstica adicional a la medición de los niveles de Lp(a), por lo que no se considera necesaria fuera de estudios de investigación108.
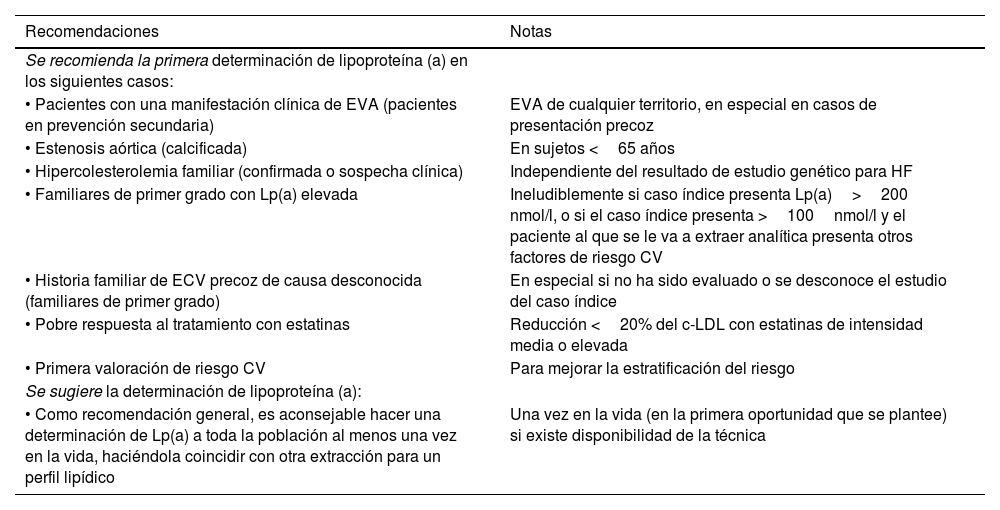
En la tabla 2 se resumen las recomendaciones y sugerencias de realizar una primera determinación de Lp(a) de la SEA.
Recomendaciones para la primera determinación de Lp(a)
| Recomendaciones | Notas |
|---|---|
| Se recomienda la primera determinación de lipoproteína (a) en los siguientes casos: | |
| • Pacientes con una manifestación clínica de EVA (pacientes en prevención secundaria) | EVA de cualquier territorio, en especial en casos de presentación precoz |
| • Estenosis aórtica (calcificada) | En sujetos <65 años |
| • Hipercolesterolemia familiar (confirmada o sospecha clínica) | Independiente del resultado de estudio genético para HF |
| • Familiares de primer grado con Lp(a) elevada | Ineludiblemente si caso índice presenta Lp(a)>200 nmol/l, o si el caso índice presenta >100nmol/l y el paciente al que se le va a extraer analítica presenta otros factores de riesgo CV |
| • Historia familiar de ECV precoz de causa desconocida (familiares de primer grado) | En especial si no ha sido evaluado o se desconoce el estudio del caso índice |
| • Pobre respuesta al tratamiento con estatinas | Reducción <20% del c-LDL con estatinas de intensidad media o elevada |
| • Primera valoración de riesgo CV | Para mejorar la estratificación del riesgo |
| Se sugiere la determinación de lipoproteína (a): | |
| • Como recomendación general, es aconsejable hacer una determinación de Lp(a) a toda la población al menos una vez en la vida, haciéndola coincidir con otra extracción para un perfil lipídico | Una vez en la vida (en la primera oportunidad que se plantee) si existe disponibilidad de la técnica |
c-LDL: concentración de colesterol de lipoproteínas de baja densidad; CV: cardiovascular; ECV: enfermedad cardiovascular; EVA: enfermedad vascular aterosclerótica; HF: hipercolesterolemia familiar; Lp(a): lipoproteína (a).
En general, dada la estabilidad de los niveles de Lp(a) a lo largo de la vida, no es necesario repetir la determinación de los niveles de Lp(a) en reiteradas ocasiones11,106 Sin embargo, es razonable plantar una nueva determinación en circunstancias clínicas en las que pueden existir cambios importantes en los niveles de Lp(a) (material adicional, tabla S1). Los cambios más acusados pueden ocurrir en la insuficiencia renal avanzada (en particular en la diálisis peritoneal) y el síndrome nefrótico15,100, en cuyo caso está más justificada una nueva determinación. En el resto de situaciones clínicas, los cambios son modestos con muy discreta repercusión en la estimación del riesgo CV, por lo que no se considera necesario en general la medición repetida de Lp(a). En caso de valores fronterizos puede considerarse una nueva determinación si la anterior fue realizada en la infancia, embarazo, disfunción hepatocelular, en situaciones de hiper/ hipotiroidismo franco, sepsis o procesos inflamatorios agudos importantes y tras la menopausia11.
Por último, parece evidente que la repetición de la determinación de Lp(a) será obligada para evaluar la respuesta al tratamiento cuando este esté disponible. Aunque en el momento actual, los iPCSK9 son los únicos fármacos hipolipemiantes de uso común con un efecto relevante sobre Lp(a) (reducción aproximada del 20%)109, existe un importante desarrollo farmacológico en marcha con medicamentos dirigidos específicamente a descender su concentración, como se desarrollará en el capítulo correspondiente del presente documento.
Estas recomendaciones y sugerencias se resumen en el material adicional, tabla S1.
La importancia de incluir Lp(a) en la estimación del riesgo CVEvaluación del riesgo conferido de Lp(a): umbral versus tasa de riesgo continuoLa relación entre la Lp(a) y el riesgo de ECV se ha explorado desde 2 puntos de vista metodológicos. Por una parte, intentando identificar determinados puntos de corte o umbrales que distingan categorías de riesgo, como se ha realizado previamente con otras partículas lipídicas, como el c-LDL, y, por otra parte, evaluando si existe una relación proporcional continua.
En la primera aproximación, se ha evaluado el riesgo conferido por la elevación de la Lp(a), definiendo distintas concentraciones umbrales o categóricas. En el Copenhagen City Heart Study82 se comprobó que a partir del percentil 66 de los niveles de Lp(a) de 7.524 sujetos (equivalente aproximadamente a 30mg/dl), hay un incremento significativo del riesgo de infarto de miocardio durante un seguimiento de 16 años.
En la segunda aproximación, se ha analizado la relación continua entre la Lp(a) y el riesgo CV: uno de los principales estudios por su tamaño muestral es el procedente del UKBiobank44 con 460.506 sujetos de mediana edad (40 a 69 años) con un seguimiento de 11,2 años. En dicho estudio se ha apreciado que, en el total de la cohorte, cada incremento de 50nmol/l en la concentración de Lp(a) comportó un aumento del riesgo de ECV del 11% (HR: 1,11 para EAC e ictus isquémico) en el rango estudiado de Lp(a) de hasta 400nmol/l. Esta cifra varió levemente según la toma de estatinas, la historia de eventos previos y la edad, y se mantuvo para ambos sexos y distintas concentraciones de colesterol LDL.
Aunque hay evidencia suficiente para mantener que la relación entre niveles de Lp(a) y riesgo de ECV es continua, la consideración de los umbrales o puntos de corte se ha asumido por motivos prácticos, de forma similar a los puntos de corte de riesgo de ECV que definen las distintas categorías de riesgo basadas en c-LDL.
Es evidente, de todas formas, que, si bien los umbrales determinan distintas «categorías», el médico debe conocer la existencia de esta asociación continua de Lp(a) y riesgo CV, y adaptar el esfuerzo terapéutico en consecuencia.
Calculadoras de riesgo según Lp(a)De los múltiples estudios que han valorado el impacto de los niveles elevados de Lp(a) en el riesgo de ECV, cabe destacar el basado en el UK-Biobank44, del que se ha construido una calculadora disponible online que permite estimar el riesgo futuro de cardiopatía isquémica e ictus, hasta los 80 años, en base a las siguientes variables: sexo, edad, colesterol total, c-HDL, c-LDL, presión arterial sistólica, tratamiento de la hipertensión arterial, índice de masa corporal, presencia de diabetes, tabaquismo, extabaquismo y antecedentes familiares110. Una vez estimado el riesgo se puede ajustar a los niveles de Lp(a) sabiendo que por cada incremento en 50nmol/l el riesgo se multiplica por 1,11. Según los autores de dicha página web, el cálculo del riesgo se basa en una modalidad de inteligencia artificial denominada Inteligencia Artificial Causal, generada gracias a la participación de la empresa «DeepCausal AI» cuya página web no tiene más contenido actualmente que el logo (https://deepcausalai.org/) en el momento de redactar este documento.
En una cohorte danesa40 de 8.720 participantes de población general reclutados entre 1991 y 1994 y seguidos sin pérdidas hasta 2011 valorando la incidencia de IAM y EAC, se ha calculado un modelo de regresión de riesgos proporcionales de COX para cada tipo de evento con factores tradicionales de riesgo (edad, sexo, colesterol total, colesterol HDL, presión arterial sistólica, tabaquismo y diabetes) y otros modelos a los que se han añadido los niveles de Lp(a) y/o genotipo de Lp(a) de riesgo (KIV-2) y/o polimorfismos rs3798220 y rs10455872. Se ha valorado el rendimiento diagnóstico de la incorporación de los nuevos marcadores mediante el índice de reclasificación neta (IRN) y el de discriminación integrada (IDI) así como cambios en el índice C. Para percentiles 80 [Lp(a)≥47mg/dl] y 95 [Lp(a)≥115mg/dl], los IRN para infarto de miocardio fueron del 16 y 23%, y para EAC del 3 y 6%, respectivamente. Los autores en su material suplementario aportan los coeficientes beta de los modelos de regresión, pero dichos modelos no se han validado en otras poblaciones.
En la cohorte italiana de Bruneck111 con 826 sujetos de población general a los que se les calculó el riesgo según Framingham y se añadió como factor adicional tener una concentración de Lp(a)>47mg/dl, se observó un aumento de riesgo de ECV importante (HR: 2,37). La adición de este factor, por tanto, resultó en una mejora de IRN e IDI.
También se ha examinado la mejora en la clasificación del riesgo de los pacientes añadiendo los niveles de Lp(a) al sistema SCORE. Con datos de 16.777 sujetos del European Prospective Investigation of Cancer (EPIC), en los que se calculó el riesgo con el SCORE y con la modificación incluyendo el umbral de 30mg/dl de Lp(a) (correspondiente al percentil 80 de dicha cohorte), se mejoró la discriminación diagnóstica, especialmente en el grupo de riesgo intermedio112. En dicho grupo, el IRN resultante fue del 8,73%; este hallazgo apoya la determinación de la Lp(a) sobre todo en los sujetos de riesgo intermedio para poder ajustar mejor la estrategia terapéutica.
Ninguno de los 3 estudios previos aporta los modelos completos de determinación de riesgo, por lo que, en la actualidad, el único sistema con acceso público para calcular el riesgo de ECV, incluyendo la Lp(a), es el accesible online en la dirección web https://www.lpaclinicalguidance.com.
UmbralesVarios estudios han valorado el riesgo de ECV conferido por distintos umbrales de Lp(a). Así valores de Lp(a)>120nmol/l en sujetos sin ECV suponen un riesgo medido como HR de 1,25 para la EAP, 1,40 para la EAC, 1,32 para el IAM, 1,11 para el ictus isquémico y 1,09 para la mortalidad CV87.
Varios documentos de consenso y guías de práctica clínica han recomendado distintos umbrales de riesgo en los últimos años. La National Lipid Association104 estableció que el umbral de 50mg/dl o 100nmol/l podría considerarse como un factor que incrementa el riesgo y por tanto para recomendar tratamiento con estatinas. Dicho valor corresponde al percentil 80 de la población caucásica americana. La American Heart Association y el American College of Cardiology consideran los umbrales de 50mg/dl o 125nmol/l como aquellos que definen un mayor riesgo de ECV113. El consenso del Reino Unido ha establecido los siguientes niveles de riesgo según los niveles de Lp(a): riesgo bajo de ECV para niveles entre 30 y 90nmol/l de Lp(a), riesgo moderado entre 90 y 200, riesgo alto entre 200 y 400 y riesgo muy alto para valores mayores de 400nmol/l100. La guía europea 2019 ya estableció que valores de Lp(a)>180mg/dl (430nmol/l) confieren un riesgo similar al de los pacientes con hipercolesterolemia familiar heterocigota102. La European Atherosclerosis Society en su documento de consenso de 202211 establece una zona gris entre 30 y 50mg/dl. Concentraciones menores de 30mg/dl (75nmol/l) no suponen un incremento significativo del riesgo de ECV, pero mayores de 50mg/dl (125nmol/l) suponen un incremento importante. La zona gris puede ser relevante en presencia de otros factores de riesgo. Aunque no hay una conversión directa entre mg/dl y nmol/l, algunos autores establecen un factor multiplicativo entre 2 y 2,5 en dicha conversión como se ha mencionado en la sección correspondiente.
Con estos datos podemos constatar que hay un consenso internacional en definir los niveles de Lp(a)<30mg/dl (75nmol/l) como un umbral de bajo riesgo y los niveles mayores de 50mg/dl (125nmol/l) como un umbral de riesgo incrementado. El umbral de 180mg/dl (430nmol/l) definiría un riesgo muy elevado equivalente al de la HF heterocigota.
Revisión del impacto de la concentración de la Lp(a) en el riesgo CV totalLa epidemiología de la ECV nos ha enseñado que las concentraciones séricas de Lp(a) no siguen una distribución poblacional normal, mostrando una clara desviación hacia la izquierda. Por tanto, y dado que gran parte de la población general tiene concentraciones de Lp(a) relativamente bajas, los estudios clínicos se centran fundamentalmente en el tercil superior de concentración de Lp(a), en donde se ha evidenciado un aumento superior al 20% en el riesgo de morbimortalidad CV.
En las líneas precedentes ha quedado patente que la Lp(a) es un factor de riesgo de ECV tanto en prevención primaria como secundaria de la ECV, incluso en presencia de niveles bajos de colesterol LDL. En el primer escenario clínico, las concentraciones de Lp(a) por encima del percentil 75 aumentan el riesgo de EA y de IAM; niveles superiores al percentil 90 se relacionan con un incremento del riesgo de insuficiencia cardiaca58, y solo para niveles extremos superiores al percentil 95 aumenta el riesgo de ictus isquémico y de mortalidad CV41,60,83. En el campo de la prevención secundaria, 3 metaanálisis han relacionado la Lp(a) elevada con un mayor riesgo de episodios CVs graves, aunque cabe mencionar la existencia de una cierta heterogeneidad entre estudios46,48,114. En los pacientes con ECV establecida del Copenhagen General Population Study47 y del AIM-HIGH115, la Lp(a) mostró una asociación significativa con la recurrencia de episodios coronarios graves. Asimismo, en un reciente metaanálisis con datos individualizados de 7 ensayos aleatorizados de intervención con estatinas en prevención primaria y secundaria, controlados con placebo, la Lp(a) basal elevada se relacionó de forma independiente y casi lineal con el riesgo de ECV116. Por otra parte, no debemos olvidar que concentraciones muy altas de Lp(a) (>180mg/dl [>430nmol/l]) identifican a sujetos con un riesgo de ECV de por vida, equivalente a la HF heterocigota no tratada102.
Datos existentes sobre la incorporación de niveles de Lp(a) en estimadores de riesgo de ECVLa adición de la Lp(a) a los algoritmos de predicción del riesgo de ECV ha arrojado efectos variables, aunque en general se puede concluir que, como acontece con gran parte de los biomarcadores, mejora de forma marginal la discriminación de riesgo. En este sentido, un estudio con más de 28.000 mujeres procedentes del Women's Health Initiative, Women's Health Study y Justification for Use of Statins in Prevention (JUPITER), la Lp(a) se asoció con la ECV solo en aquellas con una colesterolemia basal mayor de 220mg/dl, y la mejora en la predicción fue mínima117.
La reclasificación de pacientes mediante la cuantificación de Lp(a) se ha abordado en 2 estudios con seguimientos prospectivos de 15 y 6 años111,118. La adición de Lp(a) reclasificó entre el 15 y el 40% de los pacientes como de alto o bajo riesgo CV. Más recientemente, Delabays et al.119 han demostrado que, en un país de baja mortalidad CV como Suiza, la adición de la Lp(a) al modelo SCORE refinó la prevención CV, particularmente en los individuos de riesgo intermedio, con una mejora del 11,4% en la reclasificación.
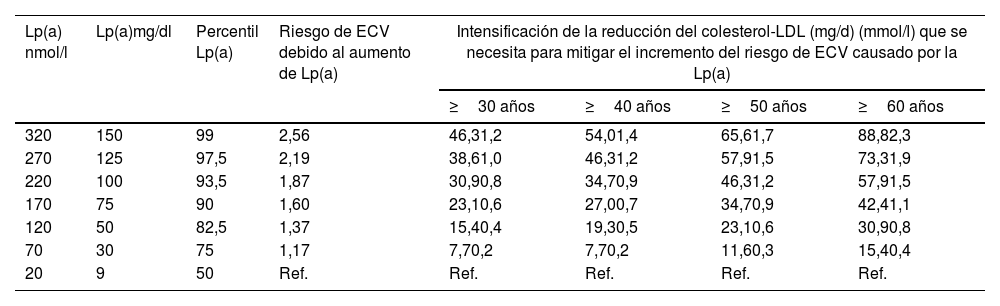
En base a ello, el consenso más reciente de la EAS11 sobre Lp(a) propone establecer qué concentraciones de Lp(a) suponen un incremento de riesgo para un sujeto determinado teniendo en cuenta su riesgo CV global. Según datos obtenidos del UK Biobank, una concentración de Lp(a) de 50mg/dl (115nmol/l) incrementa el riesgo CV absoluto de una persona con un riesgo CV global basal del 10% a lo largo de la vida en un 4% y hasta en 10% en una persona con un riesgo CV basal del 25%. Por su parte, un valor de Lp(a) de 100mg/dl (230nmol/l) aumentaría el riesgo absoluto a lo largo de la vida en un 9,5 y 24%, respectivamente11. Es decir que el impacto sobre el riesgo CV de las concentraciones de Lp(a) está condicionado también por el riesgo global basal del individuo, por lo que se requiere una actitud clínica personalizada en base a las características de la persona afecta, recomendándose, para una misma concentración de Lp(a), un manejo más intenso de los factores de riesgo en pacientes que ya tenían un riesgo elevado.
Finalmente, la aplicación de la nueva calculadora de riesgo Lp(a) propuesta por el documento de consenso de la European Atherosclerosis Society tal y como se señala en el apartado «Calculadoras de riesgo según Lp(a)», pone de relieve los siguientes aspectos. En primer lugar, hay una considerable infraestimación del riesgo de ECV si la concentración de Lp(a) es elevada y no se tiene en cuenta. En segundo lugar, la intervención sobre factores de riesgo como el colesterol c-LDL y la presión arterial, puede mitigar al menos parte del riesgo global, aunque no se modifiquen los niveles de Lp(a). Además, dado que la concentración de Lp(a) está determinada genéticamente, sus niveles permanecen estables a lo largo del tiempo para la mayor parte de la población. Por tanto, en los individuos con concentraciones elevadas de Lp(a) su carga a lo largo de la vida será muy importante.
En conclusión, aunque no existe un consenso sobre cómo incorporar el riesgo atribuible a la elevación de Lp(a) a los modelos convencionales de estimación de riesgo, la determinación de los niveles de Lp(a) puede contribuir a refinar la estimación del riesgo CV al menos semi-cuantitativamente, con una zona gris (30-50mg/dL), riesgo aumentado moderado (≈50-100mg/dl), alto (100-200mg/dl) y muy alto (>200mg/dl)
Recomendaciones para la reestimación de riesgo CV con Lp(a)Como se ha mencionado previamente, no disponemos de estudios epidemiológicos en nuestro medio que nos permitan estimar con confianza el riesgo adicional de nuestros pacientes con distintos niveles de Lp(a). Si a ello añadimos la falta de estandarización de los métodos de determinación de Lp(a) no es posible ofrecer una aproximación cuantitativa estricta. Sin embargo, es razonable atribuir un aumento del riesgo vascular similar al estimado en la población caucásica del UK Biobank y usarla como factor corrector del riesgo vascular estimado con las tablas de prevención primaria (SCORE2, SCORE-OP)11,100. El factor corrector nos permite modificar la estimación de riesgo y por tanto plantear objetivos terapéuticos de control de c-LDL acordes al riesgo recalculado.
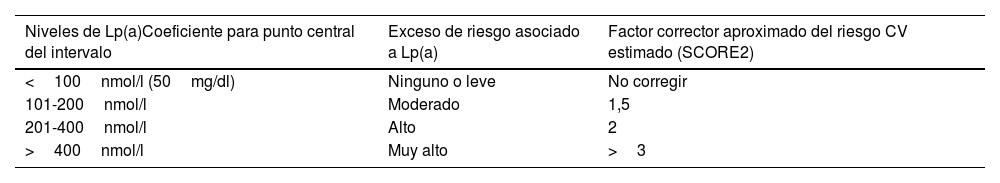
Atendiendo a la información anterior, en la tabla 3 se ofrece una aproximación para realizar una reevaluación semicuantitativa del riesgo CV añadiendo la Lp(a), usando coeficientes de corrección calculados a partir de los cambios de riesgo CV estimados en el UK Biobank para caucásicos con las escalas británicas de riesgo «a lo largo de la vida». Como se indica en el apartado correspondiente, no es posible realizar una conversión única entre unidades molares y masa de Lp(a) por lo que los coeficientes de corrección deben emplearse con precaución. La tabla es una aproximación semicuantitativa, de modo que los coeficientes son una estimación de los valores medios de los intervalos descritos, pudiendo ser ajustados con cierta mayor precisión tal y como se muestra en el material adicional, tabla S2.
Modelo simplificado calculado a partir de los cambios de riesgo CV estimados en el UK Biobank para caucásicos con las escalas de riesgo CV británicas de riesgo «a lo largo de la vida»
| Niveles de Lp(a)Coeficiente para punto central del intervalo | Exceso de riesgo asociado a Lp(a) | Factor corrector aproximado del riesgo CV estimado (SCORE2) |
|---|---|---|
| <100nmol/l (50mg/dl) | Ninguno o leve | No corregir |
| 101-200nmol/l | Moderado | 1,5 |
| 201-400nmol/l | Alto | 2 |
| >400nmol/l | Muy alto | >3 |
Se ofrece la escala propuesta para Lp(a) en unidades molares y las unidades en masa reportadas en el consenso original. Como se indica en el apartado correspondiente, no es posible realizar una conversión única entre unidades molares y masa de Lp(a) por lo que los coeficientes de corrección deben emplearse con precaución. El riesgo CV estimado para incluir el exceso de riesgo por Lp(a) es la multiplicación de la estimación del riesgo (v. gr. SCORE2) por el coeficiente de corrección de la tabla. Se aporta un modelo más detallado en el material suplementario de este consenso.
CV: cardiovascular; Lp(a): lipoproteína (a).
En los pacientes en prevención secundaria, la Lp(a) elevada confiere también un riesgo adicional por lo que puede ser tenida en cuenta para la identificación de pacientes de riesgo CV extremo en los que pueden plantearse objetivos más ambiciosos de control de c-LDL (<40mg/dl) y el uso precoz de tratamientos especialmente intensivos, como los iPCSK9, tal como se recoge en las recomendaciones de la SEA107.
Como se ha mencionado en el apartado correspondiente, no disponemos en el momento actual tratamientos que hayan demostrado reducción de complicaciones cardiovasculares mediante tratamientos dirigidos a la reducción de Lp(a). Por este motivo debe animarse a los clínicos y pacientes a la participación en ensayos clínicos que permitan ofrecer en los próximos años un tratamiento basado en la mejor evidencia científica.
Por último, una limitada evidencia clínica120 sugiere que el uso de aféresis de lipoproteínas en pacientes con EAC puede mejorar la evolución clínica de los mismos. No existe una indicación asentada para esta modalidad terapéutica, pudiendo plantearse de modo individualizado en pacientes con niveles extremadamente elevados de Lp(a) con ECV establecida, o incluso inferiores en caso de progresión de lesiones vasculares a pesar de control adecuado del resto de factores de riesgo CV.
Finalmente, hay que recordar que independientemente del riesgo CV calculado sin tener en cuenta la Lp(a), distintas guías europeas ya se han pronunciado con relación al aumento de riesgo conferido únicamente por la exposición a niveles muy altos de Lp(a). Así, los pacientes con niveles extremos de Lp(a), percentil mayor del 99% (400nmol/l; 200mg/dl) deberían considerarse per se cómo pacientes de riesgo CV alto de partida, incluso en ausencia de otros factores de riesgo y por tanto candidatos a plantear reducción del c-LDL por debajo de 70mg/dl100,102.
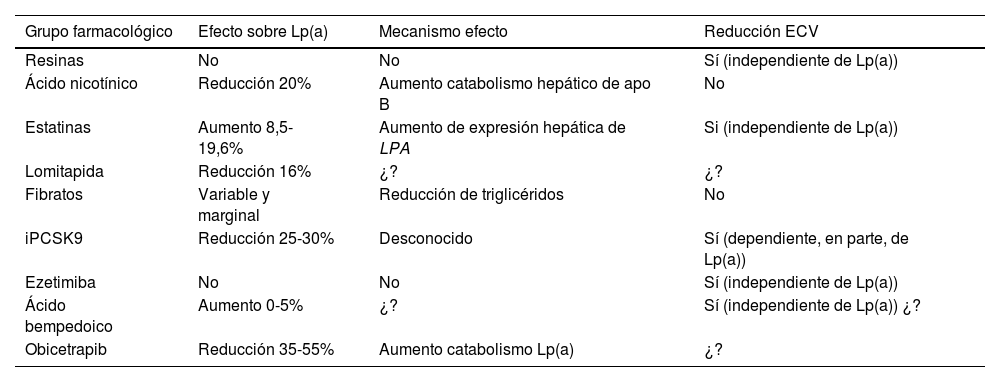
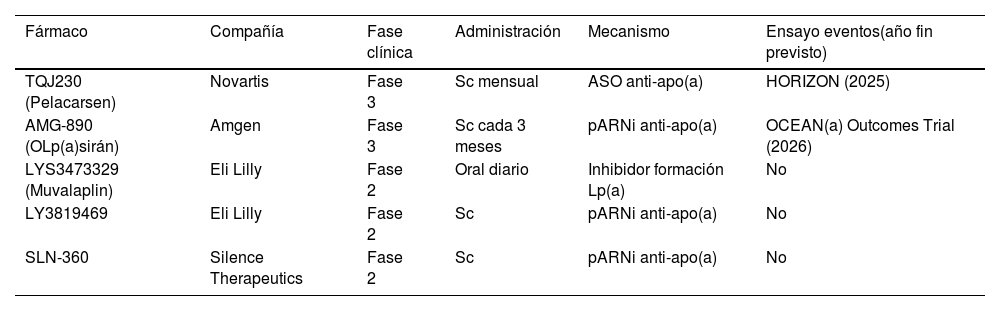
Dianas y estrategias terapéuticas para reducir los niveles de Lp(a)Aproximación farmacológica actualEn la actualidad no existe ningún tratamiento farmacológico aprobado para reducir específicamente las concentraciones plasmáticas elevadas de Lp(a), y de confirmarse en los estudios de intervención en marcha los beneficios esperados en base a la evidencia observacional, será una necesidad clínica y uno de los grandes retos en la prevención y tratamiento de la arteriosclerosis. Los fármacos hipolipemiantes actuales tienen un efecto neutro o clínicamente poco relevante en la concentración de Lp(a) (tabla 4). Sin embargo, en los últimos años, nuevas terapias, algunas de ellas en las últimas fases de su desarrollo, auguran una esperanza en el manejo de la hiper Lp(a).
Fármacos comercializados y su relación con las concentraciones de Lp(a)
| Grupo farmacológico | Efecto sobre Lp(a) | Mecanismo efecto | Reducción ECV |
|---|---|---|---|
| Resinas | No | No | Sí (independiente de Lp(a)) |
| Ácido nicotínico | Reducción 20% | Aumento catabolismo hepático de apo B | No |
| Estatinas | Aumento 8,5-19,6% | Aumento de expresión hepática de LPA | Si (independiente de Lp(a)) |
| Lomitapida | Reducción 16% | ¿? | ¿? |
| Fibratos | Variable y marginal | Reducción de triglicéridos | No |
| iPCSK9 | Reducción 25-30% | Desconocido | Sí (dependiente, en parte, de Lp(a)) |
| Ezetimiba | No | No | Sí (independiente de Lp(a)) |
| Ácido bempedoico | Aumento 0-5% | ¿? | Sí (independiente de Lp(a)) ¿? |
| Obicetrapib | Reducción 35-55% | Aumento catabolismo Lp(a) | ¿? |
apo B: apolipoproteína B; ECV: enfermedad cardiovascular; Lp(a): lipoproteína(a); PCSK9: proproteína convertasa subtilisina/kexina tipo 9; ¿?: dudosa.
A diferencia de lo que ocurre con otras partículas lipoproteicas, como los niveles de c-LDL o los triglicéridos, la concentración plasmática de Lp(a) no se modifican de forma clínicamente relevante con los cambios en la dieta y el estilo de vida saludable121. El efecto de los hipolipemiantes actuales sobre la concentración de Lp(a) se resume en la tabla 4. Si bien alguno de estos fármacos reduce de forma significativa los niveles de Lp(a), no existen datos sobre el posible beneficio CV mediado por este descenso.
Las resinas no han demostrado que modifiquen los niveles de Lp(a)122. La niacina produce una reducción en los niveles de Lp(a) en torno a un 20% 123,124. Estudios previos indican que el tratamiento con estatinas puede aumentar porcentualmente los niveles de Lp(a). En este contexto, un metaanálisis reciente, incluyendo 39 estudios y 24.448 participantes, muestra que el tratamiento con estatinas no modificó el riesgo de ECV asociado al ligero aumento de los niveles de Lp(a), siendo por tanto dicho cambio irrelevante desde el punto de vista clínico125. Dado el efecto protector del tratamiento reductor del colesterol LDL es proporcional al riesgo CV basal, la inclusión de Lp(a) en la estimación de riesgo puede identificar a pacientes con mayor beneficio potencial por su mayor riesgo absoluto. Este hecho apoya que en pacientes con riesgo CV moderado o fronterizo, y niveles de Lp(a) elevado (>50mg/dl), se deba recalcular su riesgo y valorar el inicio de tratamiento con hipolipemiante, en principio con estatinas104 y considerar la intensificación de este en pacientes con riesgo alto o muy alto. En otras palabras, la elevación de Lp(a) puede aconsejar alcanzar objetivos de control de c-LDL más ambiciosos. La lomitapida, cuya indicación actual única es la HF homocigota, reduce los niveles de Lp(a) en sujetos con hipercolesterolemia126,127. Entre los fibratos el efecto es muy variable y depende de la concentración basal de triglicéridos14. En un metaanálisis de 7 ensayos de monoterapia con ezetimiba, esta redujo significativamente los niveles de Lp(a) en un 7,1%128. Por el contrario, en otro metaanálisis, la ezetimiba, tanto en monoterapia frente a placebo o en combinación con estatina frente a estatina sola, no modificó significativamente las concentraciones de Lp(a)129. Más recientemente, en un ensayo de fase 2, el ácido bempedoico no redujo de forma significativa los niveles de Lp(a)130,131. Del mismo modo, la combinación de ácido bempedoico con evolocumab no fue superior a la monoterapia con evolocumab en cuanto a la variación de la Lp(a) con respecto al valor basal132. En definitiva, todos estos fármacos hipolipemiantes, bien por falta de eficacia, ausencia de un beneficio clínico relevante, o por los propios efectos secundarios de algunas de estas moléculas, no son herramientas que estén indicadas para reducir los niveles elevados de Lp(a)133.
Los iPCSK9 (evolocumab y alirocumab) han mostrado un marcado efecto sobre los niveles de Lp(a) con una reducción media del 26,7% (IC 95%: −29,5 a −23,9%) con una heterogeneidad significativa en relación con el comparador y la duración del tratamiento134.
Los análisis de subgrupos de los estudios Odyssey y Fourier muestran un beneficio absoluto de reducción de eventos CVs superior en pacientes con Lp(a) elevada tras tratamiento con alirocumab y evolocumab, respectivamente. Si bien parte del beneficio puede atribuirse al mayor riesgo absoluto de los pacientes con Lp(a) elevada, análisis post hoc sugieren la posibilidad de un beneficio adicional ligado a la reducción de Lp(a) per se, independiente del efecto sobre el c-LDL. El diseño de los estudios y la naturaleza post hoc de estos análisis no permiten extraer una conclusión definitiva al respecto135,136.
Inclisirán, un pequeño ARN de interferencia (pARNi) dirigido a la PCSK9 intracelular, ha demostrado reducir los niveles de Lp(a) en aproximadamente un 20%137. El beneficio clínico de este hallazgo se desconoce en la actualidad, aunque se podría hipotetizar que sea similar a la de los iPCSK9. Por último, cabe señalar que Obicetrapib, un inhibidor selectivo de la proteína de transferencia de éster de colesterol (CETP) en fase de desarrollo clínico, a dosis de 5 y 10mg ha demostrado una reducción de la Lp(a) en un 33,8 y un 56,5%, respectivamente138.
Los fracasos en algunos ensayos clínicos con fármacos con presumible efecto favorable sobre la ECV por las mejoras en el perfil lipídico aterogénico (incluida reducción de Lp(a)) con niacina y con inhibidores de CETP, obligan a la precaución antes de asumir beneficios CVs de los fármacos reductores de Lp(a) mientras no se hayan desarrollado adecuados ensayos clínicos que evalúen específicamente objetivos clínicos relevantes, no simplemente ‘mejoras’ del perfil lipídico.
AféresisHasta ahora, la intervención más eficaz para reducir las concentraciones de Lp(a) es la aféresis de lipoproteínas139. La eficacia de dicha intervención es notable, reduciendo los niveles de Lp(a) entre un 70-80%, aunque posteriormente aumentan alcanzando una reducción media del intervalo de Lp(a) del 25-40%, dependiendo del curso y de los niveles basales de Lp(a)139.
Un estudio simple ciego aleatorizado cruzado en 20 pacientes con angina refractaria y Lp(a) elevada sometidos a aféresis de lipoproteínas durante 3 meses sugiere que esta intervención puede mejorar la perfusión miocárdica y mejorar el control de la angina140.
Se está llevando a cabo un ECA denominado MultiSELECt en el que se compara la aféresis de Lp(a) semanal con el tratamiento hipolipemiante máximo tolerado en aproximadamente 1.000 pacientes de entre 18 y 70 años con niveles de Lp(a)>120 nmol/l o superiores, niveles de c-LDL<100mg/dl y ECV. El objetivo final planteado es explorar el beneficio clínico de la aféresis de Lp(a) sobre MACE120.
Nuevas terapias en desarrolloUn enfoque completamente diferente para la reducción de proteínas abundantes implica el bloqueo de su producción mediante la inhibición de la traducción de su ARNm. Las 2 clases principales de fármacos dirigidos a ARN desarrollados para este propósito son los oligonucleótidos antisentido (ASO) monocatenarios y los pARNI (tabla 5). Ambas estrategias comparten un mecanismo de acción similar: tras la administración parenteral y su introducción en el hepatocito, se unen a una secuencia específica del ARNm de interés, a nivel nuclear para los ASO y en el citoplama en el caso de los pARNi. Esta interacción conduce a la degradación del ARNm objetivo y, por lo tanto, disminución de la traducción de la proteína codificada. Una de las principales ventajas de los fármacos basados en ARN es la posibilidad de dirigirse con elevada especificidad contra proteínas con altas concentraciones plasmáticas como la apo(a), componente de la partícula Lp(a)141. Pelacarsen es un ASO sintético de 20 nucleótidos, conjugado con GalNAc3 y dirigido al ARNm de la apo(a), que ha demostrado en ensayos en fase 1 y 2 de hasta el 80%141,142. Se está evaluando el impacto de 80mg mensuales de Pelacarsen sobre la reducción del MACE en el ensayo HORIZON (NCT04023552), un ECA de fase 3 que incluye a más de 7.000 pacientes con ECV y Lp(a)>70mg/dl, con el fin de comparar su eficacia y seguridad. Los resultados se esperan para 2025143. OLp(a)sirán, es un pARNi que interrumpe la producción de apo(a) a través de la degradación del ARN mensajero de apo(a), lo que impide el ensamblaje de la Lp(a) en el hepatocito. En el desarrollo preclínico ha demostrado una reducción del Lp(a) de hasta el 98% en pacientes con niveles> 70nmol/l sin acontecimientos adversos graves144. En un ECA de fase 2, OCEAN(a) DOSE (OLp(a)siran Trials of ECV Events and LipoproteiN(a) Reduction) 281 sujetos con ECV y niveles de Lp(a)>150nmol/l asignados a recibir OLp(a)siran (10mg/cada 12 semanas, 75mg/cada 12 semanas, 225mg/cada 12 semanas o 225mg/cada 24 semanas) obtuvieron reducciones significativas de los niveles de Lp(a) (70,5, 97,4, 101,1 y 100,5%, respectivamente)145. El ECA de fase 3, OLp(a)siran Trials of ECV Events and Lipoprotein(a) Reduction (OCEAN(a)) - Outcomes Trial (NCT05581303)146 se encuentra en fase de reclutamiento activo. Otro pARNi, SLN360 dirigido a bloquear la apo(a), ha obtenido reducciones entre el 46% y el 98% con terapia administrada por vía subcutánea en una dosis única de: 30, 100, 300 o 600mg en pacientes con Lp(a)>150 nmol/l147. SLN360 también tiene otro ECA de fase 2 en curso (Estudio para investigar la seguridad, tolerabilidad, PK y respuesta a la PD de SLN360 en sujetos con lipoproteína(a) elevada; NCT04606602) pendiente de divulgación de resultados148. En el estudio ALP(A)CA, el objetivo principal es determinar la eficacia y la seguridad de LY3819469 en adultos con Lp(a) elevada149. Por último, la compañía Eli Lilly tiene una pequeña molécula de administración diaria oral (LYS3473329 (Muvalaplin)), que inhibe la formación de Lp(a) y con resultados prometedores. Las futuras direcciones con terapias orales e incluso con tratamientos de edición genética con nanopartículas capaces de modular la transcripción y traducción de la Lp(a) son el primer paso hacia el control definitivo del riesgo CV residual favorecido por esta Lp150. Todo esto se resume en la tabla 5.
Fármacos en estudio en la reducción de Lp(a)
| Fármaco | Compañía | Fase clínica | Administración | Mecanismo | Ensayo eventos(año fin previsto) |
|---|---|---|---|---|---|
| TQJ230 (Pelacarsen) | Novartis | Fase 3 | Sc mensual | ASO anti-apo(a) | HORIZON (2025) |
| AMG-890 (OLp(a)sirán) | Amgen | Fase 3 | Sc cada 3 meses | pARNi anti-apo(a) | OCEAN(a) Outcomes Trial (2026) |
| LYS3473329 (Muvalaplin) | Eli Lilly | Fase 2 | Oral diario | Inhibidor formación Lp(a) | No |
| LY3819469 | Eli Lilly | Fase 2 | Sc | pARNi anti-apo(a) | No |
| SLN-360 | Silence Therapeutics | Fase 2 | Sc | pARNi anti-apo(a) | No |
apo (a): poproteína (a); ASO: oligonucleótidos antisentido; Lp(a): lipoproteína(a); pARNi: pequeño ARN bicatenarios de interferencia; Sc: subcutáneo.
Otros ensayos clínicos de interés actualmente en desarrollo se muestran en el material adicional, tabla S3.
Beneficios esperables de la reducción de Lp(a)No disponemos de ningún estudio que haya demostrado el beneficio clínico de la reducción de la Lp(a). Esto no impide que podamos realizar una estimación del beneficio esperable de la reducción de Lp(a) con fármacos a partir del riesgo atribuible a la Lp(a) en estudios epidemiológicos y la reducción de esta con diferentes intervenciones. En este sentido, cabe destacar el ejercicio realizado a partir de estudios de aleatorización mendeliana en paralelo con los efectos conocidos de la reducción de LDL. Burgess et al. han estimado que sería precisa una reducción de alrededor de 100mg/dl de Lp(a) para obtener un beneficio similar al alcanzado con un descenso de c-LDL de 1mmol/l (≈40mg/dl)151. Este ejercicio, por teórico que sea, ofrece perspectivas de interés. En primer lugar, nos da una estimación de cómo un tratamiento intensivo de reducción de LDL podría contrarrestar el «exceso» de riesgo atribuible al aumento de Lp(a). Podemos comprobar cómo una reducción fácilmente accesible de c-LDL (v. gr. 40% reducción de un paciente con LDL=100mg/dl) exigiría una reducción mucho más intensa (v. gr. reducción de>60% de Lp(a) en un paciente con Lp(a) de 150mg/dl) para una reducción de riesgo equivalente. Por tanto, las reducciones de Lp(a) deben ser especialmente intensas para lograr un beneficio clínico significativo. Las reducciones modestas (v. gr. 20% obtenido con iPCSK9) tendrían por tanto un beneficio limitado. Afortunadamente, los estudios en fase 2 con ASO y pARNi indican que se pueden obtener reducciones importantes (>75%) con tratamientos bien tolerados. El balance riesgo/beneficio en términos de eventos CV y seguridad debe esperar a la conclusión de ensayos cínicos adecuadamente diseñados. Debe pues animarse a los pacientes con Lp(a) elevada y médicos que los atienden a participar en estos ensayos para poder ofrecer un tratamiento adecuado en los próximos años.
Manejo de pacientes con Lp(a) alta: ¿Por qué aumentar el control del resto de factores de riesgo de ECV en personas con niveles altos de Lp(a)?MotivaciónComo se ha mencionado con anterioridad, la asociación de concentraciones plasmáticas elevadas de Lp(a) e incremento del riesgo tanto de EVA como de lesión valvular aórtica ha quedado bien establecida. En las recomendaciones de la EAS sobre Lp(a) publicadas en 2010152, se establecía, en base a los datos del Copenhagen General Population Study82, un único nivel umbral de riesgo, 50mg/dl (115nmol/l), a partir del cual se consideraba necesario reconsiderar el riesgo global de los pacientes. Los estándares SEA 2024 para el control global del riesgo CV153 recogen también este umbral para señalar el riesgo adicional atribuido por las elevadas concentraciones de Lp(a). Los datos actuales sugieren que el incremento de riesgo proporcionado por las concentraciones de Lp(a) es directo y continuo154.
Prevalencia de Lp(a) elevada en EspañaActualmente, en España, hay poca evidencia sobre la prevalencia de Lp(a) elevada. En un estudio realizado en población de las Islas Canarias en 2012 se realizó un cribado sobre 1.030 personas donde se encontró una prevalencia de valores superiores a 47mg/dl del 13,7%155. Existen otros estudios donde se ha evaluado la prevalencia de la Lp(a) elevada en los resultados de las analíticas solicitadas en práctica clínica. En general, la prevalencia de Lp(a) elevada (>50mg/dl) en estos estudios es superior (25-30%), si bien, por lo general, los datos provienen de poblaciones seleccionadas por alto riesgo CV o sospecha de Lp(a) elevada (familiares de personas con Lp(a) elevada, personas en prevención secundaria o alto riesgo CV, etc) por lo que se puede estar incurriendo en una sobreestimación por sesgo de selección156–158.
Disminución del riesgo CV según el riesgo realEn el último consenso europeo sobre la Lp(a) se sugiere cuánto debería ser la reducción adicional del c-LDL para reducir el riesgo de ECV global debido al aumento de la concentración de Lp(a), basado en los datos del UK Biobank11 (tabla 6). Dado que este cálculo se ha establecido en base al riesgo CV a lo largo de la vida, el factor edad es también muy importante, al representar diferentes períodos de exposición a las concentraciones de Lp(a) elevadas. Así, en un paciente con una Lp(a) de 100mg/dl (unos 220nmol/l), para anular el riesgo de ECV atribuido a dicha concentración de Lp(a), tendría que reducirse el c-LDL unos 31mg/dl (0,8nmol/l) si tuviera 30 años o unos 58mg/dl (1,5nmol/l) si tuviera 60 años (tabla 6)11. Por lo tanto, esta tabla da una información práctica para estimar cuanto intensificar el tratamiento hipolipemiante según la concentración de Lp(a) de nuestros pacientes. Como se ha comentado anteriormente (apartado «Recomendaciones para la reestimación de riesgo CV con Lp(a)»), una Lp(a) extrema (mayor al percentil 99%; 400nm/l; >200mg/dl) ha sido identificada en varias guías europeas como equivalente a HF en objetivo de LDL, y por tanto, debería reducirse LDL a <70mg/dl100,102.
Incremento de riesgo de ECV según las concentraciones de Lp(a) y grado de reducción del colesterol-LDL para mitigar dicho aumento, dependiendo de la edad del paciente
| Lp(a) nmol/l | Lp(a)mg/dl | Percentil Lp(a) | Riesgo de ECV debido al aumento de Lp(a) | Intensificación de la reducción del colesterol-LDL (mg/d) (mmol/l) que se necesita para mitigar el incremento del riesgo de ECV causado por la Lp(a) | |||
|---|---|---|---|---|---|---|---|
| ≥30 años | ≥40 años | ≥50 años | ≥60 años | ||||
| 320 | 150 | 99 | 2,56 | 46,31,2 | 54,01,4 | 65,61,7 | 88,82,3 |
| 270 | 125 | 97,5 | 2,19 | 38,61,0 | 46,31,2 | 57,91,5 | 73,31,9 |
| 220 | 100 | 93,5 | 1,87 | 30,90,8 | 34,70,9 | 46,31,2 | 57,91,5 |
| 170 | 75 | 90 | 1,60 | 23,10,6 | 27,00,7 | 34,70,9 | 42,41,1 |
| 120 | 50 | 82,5 | 1,37 | 15,40,4 | 19,30,5 | 23,10,6 | 30,90,8 |
| 70 | 30 | 75 | 1,17 | 7,70,2 | 7,70,2 | 11,60,3 | 15,40,4 |
| 20 | 9 | 50 | Ref. | Ref. | Ref. | Ref. | Ref. |
Lp(a): lipoproteína(a).
Fuente: tomada y modificada de Kronenberg et al. 11. Se ha añadido una columna con la conversión aproximada de la concentración de Lp(a) de nmol/l a mg/dl y la conversión de la concentración de colesterol-LDL de mmol/l a mg/dl.
En el estudio EPIC-Norfolk se evaluó como las personas con Lp(a) elevada podrían reducir su riesgo CV controlando otros factores de riesgo159. Los 14.000 participantes fueron estratificados según la puntuación en 7 parámetros de salud CV (índice de masa corporal, dieta sana, actividad física, tabaquismo, presión arterial, diabetes y concentración de colesterol) en 3 grupos: saludable, intermedio y no saludable. Tras 11,5 años de seguimiento, entre los pacientes con concentraciones de Lp(a) ≥50mg/dl, aquellos incluidos en el grupo con puntuación de salud CV saludable tuvieron un 67% menos eventos CVs que los del grupo no saludable. Aunque la concentración de Lp(a) estaba poco influenciada por los factores de riesgo de CV, una buena salud CV podría reducir sustancialmente el riesgo asociado a unos niveles elevados de Lp(a). Por lo tanto, la falta de un tratamiento de eficacia probada para reducir específicamente la Lp(a) y la ECV no debe ser un obstáculo (sino todo lo contrario) para intensificar el tratamiento hipolipemiante, adoptar estilos de vida saludables, mantener el peso ideal, evitar el tabaquismo, controlar la presión arterial y la glucemia.
La intensificación del tratamiento hipolipemiante se debe hacer fundamentalmente con estatinas. Si bien algunos autores han descrito un ligero aumento de Lp(a) con el uso de las estatinas, un reciente metaanálisis con 39 estudios y más de 20.000 pacientes no encontró diferencias clínicamente importantes en la concentración de Lp(a) en comparación con placebo en pacientes con riesgo de ECV125. Por otra parte, los iPCSK9 además de reducir el c-LDL un 60%, disminuyen alrededor del 25% los niveles de Lp(a), siendo los pacientes con niveles basales de Lp(a) más elevados los que experimentan mayores reducciones absolutas de Lp(a) y los que tienden a obtener un mayor beneficio coronario con la inhibición de PCSK9136,160.
Como hemos comentado anteriormente, existe una página web (https://www.lpaclinicalguidance.com) en la que se puede calcular el riesgo de ECV de por vida, incluyendo los factores de riesgo clásicos y la concentración de Lp(a). Además, en esa misma página se puede estimar cuánto puede mejorar dicho riesgo para descensos concretos de c-LDL y/o presión arterial sistólica.
Por último, además de intervenir sobre los factores de riesgo en los sujetos con Lp(a) elevada podría ser interesante disminuir su riesgo trombótico con tratamiento antiagregante. En el estudio ASPREE se evaluó la utilidad de 100mg/día de aspirina frente a placebo en 19.000 sujetos mayores de 70 años en prevención primaria con resultado negativo tras 5 años de seguimiento. En casi 13.000 de estos sujetos se determinó el polimorfismo rs3798220, cuyo alelo C se asoció a concentraciones elevadas de Lp(a) y a un aumento de la incidencia de ECV en el grupo placebo. Al comparar la incidencia de ECV en los portadores de dicho alelo, aquellos que recibieron aspirina tuvieron una disminución significativa de ECV comparados con el grupo placebo161. Por lo tanto, a la espera de ensayos clínicos que confirmen estos datos, los resultados de este estudio observacional sugerirían que los sujetos en prevención primaria con Lp(a) elevada podrían beneficiarse del tratamiento preventivo con aspirina.
Conclusiones- •
Este consenso de la SEA se realiza dadas las evidencias cada vez más intensas que asocian a la Lp(a) con el riesgo CV, especialmente a partir de los dinteles de 100 y 200nmol/l o 50 y 100mg/dl.
- •
Es fundamental avanzar hacia una uniformidad en la determinación de la Lp(a), tanto en metodología como en unidades de medida.
- •
La Lp(a) tiende a mantenerse por regla general en una concentración similar a lo largo del tiempo y la mayor parte está condicionada genéticamente, por lo que las actuaciones sobre el estilo de vida tienen un impacto leve sobre su concentración.
- •
Dada su estabilidad en el tiempo, la SEA sugiere la determinación universal de Lp(a) una vez en la vida coincidiendo con la primera determinación bioquímica donde se incluya el perfil lipídico.
- •
La SEA recomienda determinar la Lp(a) de manera inmediata si no se ha realizado nunca, en presencia de ECV, estenosis aórtica, hipercolesterolemia familiar, existencia de familiares de primer grado con Lp(a) elevada o con ECV precoz de causa incierta, o en pacientes con pobre respuesta hipolipemiante al tratamiento con estatinas.
- •
Se recomienda la repetir la determinación de Lp(a) en casos específicos (ver recomendaciones en el apartado correspondiente)
- •
A falta de ensayos clínicos específicos sobre reducción de Lp(a) y eventos cardiovasculares, toda la evidencia actual indica que la Lp(a) puede ser el actor más importante aparecido en el campo de la lipidología en los últimos 50 años.
- •
La SEA recomienda la adaptación del esfuerzo terapéutico a la concentración de Lp(a) en todos los pacientes.
- •
La aparición de calculadoras de riesgo que incluyen la Lp(a), como la que existe en https://www.lpaclinicalguidance.com, puede ser de ayuda para el profesional sanitario en la estimación del riesgo global del RCV.
- •
En este mismo documento se ofrecen unas tablas para la reevaluación del riesgo CV adaptándolas a la concentración de Lp(a).
- •
El único grupo farmacológico que ha demostrado hasta la fecha cierto poder terapéutico para reducir la Lp(a) son los iPCSK9.
- •
El presente documento contiene también una tabla para consultar la intensificación del tratamiento sobre el c-LDL para contrarrestar el efecto de la Lp(a) elevada.
- •
Los resultados de estudios clínicos de reducción de Lp(a) en prevención de la ECV podrá variar la posición de la SEA.
El Dr. Delgado-Lista F.J. ha recibido honorarios por conferencias, eventos educativos y soporte para reuniones por Amgen, Novartis, Ferrer, Laboratorios Dr. Esteve, Menarini, Novo-Nordisk, Sanofi, MSD, Servier, Astra-Zeneca y Mylan.
El Dr. Mostaza J.M. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos de Almirall, Alter, Amgen, Daiichi-Sankyo, Ferrer, Novartis, Sanofi, Servier, Viatris y Ultragenix.
La Dra. Arrobas-Velilla T. ha recibido honorarios de consultoría por Amgen, Sanofi, Amarin, Daiichi y Novartis; por conferencias, presentaciones, manuscrito o eventos educativos y reuniones por Amgen, Sanofi, Mylan, Sevier, Amarin, Amryt, Daiichi y Novartis.
El Dr. Blanco Vaca J. ha recibido honorarios por conferencias de Daiichi-Sankyo y European Atherosclerosis Society, y apoyo económico para organización de cursos de Amarin, Daiichi-Sankyo, MSD, Novartis y Sanofi.
El Dr. Masana L. ha recibido honorarios por conferencias o trabajo de consultoria de Amarin, Amryt, Daiichi-Sankyo, Novartis, MSD, Sanofi, Servier, Ferrer y Ultragenix.
El Dr. Pedro-Botet J. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos, soporte para reuniones por Amarin, Amgen, Daiichi-Sankyo, Esteve, Ferrer, MSD, Novartis, Sanofi y Viatris.
El Dr. Perez-Martinez P. ha recibido honorarios por conferencias, presentaciones, o eventos educativos y reuniones por Ferrer, Novo-Nordisk, Amgen, Daiichi Sankyo, Esteve y Viatris.
El Dr. Civeira F. ha recibido honorarios por conferencias y Advisory Boards de Amgen, Sanofi, Novartis y Ferrer
El Dr. Cuende Melero J.I. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito, eventos educativos, soporte para reuniones o contratos de por MSD, Daiichi-Sankyo, Sanofi, Bayer y Almirall.
El Dr. Gómez Barrado J.J. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos, soporte para reuniones por Amarin, Amgen, Novartis, Ferrer, Daiichi-Sankyo y Sanofi.
El Dr. Lahoz C. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos, soporte para reuniones por Amarin, Alter, Novartis, Daiichi-Sankyo y Sanofi.
El Dr. Pintó X. ha recibido honorarios de consultoría, por experto testimonio, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos, soporte para reuniones de Akcea, Almirall, Amarin, Amgen, Daiichi-Sankyo, Ferrer, Menarini, Novartis, Organon, Sanofi, Sobi y Viatris.
El Dr. Suarez Tembra M. ha recibido honorarios por conferencias de Laboratorios Servier y Daichi, y ayuda para asistencia a congresos de Amgen, Servier, Daichi y Alter.
El Dr. Lopez-Miranda J. ha recibido honorarios de consultoría por Amgen y Sanofi, y por conferencias, presentaciones, manuscrito o eventos educativos y reuniones por Amgen, Sanofi, Novartis, Laboratorios Dr. Esteve y Laboratorios Ferrer.
El Dr. Guijarro C. ha recibido honorarios de consultoría, por conferencias, Advisory Boards, presentaciones, manuscrito o eventos educativos, por Amarin, Amgen, Daiichi-Sankyo, Ferrer, Novartis, MSD, Sanofi y Servier.