





Atherosclerosis is the main pathogenic substrate for cardiovascular diseases (CVDs). Initially categorized as a passive cholesterol storage disease, nowadays, it is considered an active process, identifying inflammation among the key players for its initiation and progression. Despite these advances, patients with CVDs are still at high risk of thrombotic events and death, urging to deepen into the molecular mechanisms underlying atherogenesis, and to identify novel diagnosis and prognosis biomarkers for their stratification. In this context, extracellular vesicles (EVs) have been postulated as an alternative in search of novel biomarkers in atherosclerotic diseases, as well as to investigate the crosstalk between the cells participating in the processes leading to arterial remodelling. EVs are nanosized lipidic particles released by most cell types in physiological and pathological conditions, that enclose lipids, proteins, and nucleic acids from parental cells reflecting their activation status. First considered cellular waste disposal systems, at present, EVs have been recognized as active effectors in a myriad of cellular processes, and as potential diagnosis and prognosis biomarkers also in CVDs. This review summarizes the role of EVs as potential biomarkers of CVDs, and their involvement into the processes leading to atherosclerosis.
La aterosclerosis es el principal sustrato patogénico de las enfermedades cardiovasculares (ECV). Considerada inicialmente como un depósito pasivo de colesterol, hoy se asume que es un proceso activo, donde la inflamación juega un papel clave en su inicio y progresión. A pesar de los avances realizados, los pacientes con ECV presentan alto riesgo de episodios trombóticos y elevada mortalidad, por lo que sigue siendo necesario profundizar en los mecanismos de la aterogénesis e identificar nuevos biomarcadores diagnósticos y pronósticos para estratificar el riesgo. En este sentido, se ha postulado que las vesículas extracelulares (EV) podrían representar nuevos biomarcadores de la enfermedad aterosclerótica, siendo de interés investigar su participación en la comunicación intercelular que favorece el remodelado arterial. Las EV son partículas lipídicas liberadas por numerosas células en condiciones fisiológicas y patológicas, que contienen lípidos, proteínas y ácidos nucleicos procedentes de las células parentales. Consideradas inicialmente como material de desecho, hoy se sabe que son efectores activos de numerosos procesos celulares, constituyendo biomarcadores potenciales con significado diagnóstico y pronóstico en la ECV. En esta revisión se sintetiza el papel de las EV como biomarcadores potenciales de las ECV y su participación en procesos que favorecen la aterosclerosis.
Atherosclerosis is a major cause of cardiovascular diseases (CVDs), predisposing patients to myocardial infarction and stroke, which are the primary causes of morbidity and mortality worldwide. An estimated 17.9 million people died from CVD in 2019, representing 32% of all global deaths. These figures are expected to grow to more than 23.6 million by 2030 due to unhealthy lifestyle changes and ageing.1,2 Atherosclerosis is a multifactorial disease, that progresses silently according to genetic factors (e.g.: familial hypercholesterolemia), age, and/or exposure to risk factors, including, dyslipidemia, hypertension, diabetes mellitus, kidney diseases or smoking.3–5 It can be latent and symptomless, but in advanced stages atherosclerosis may trigger symptoms of acute coronary syndrome, heart failure, stroke or peripheral vascular disease (PAD), that are the most common consequences of atherosclerosis.1,6 The identification of atherogenesis as an active process, rather than the passive cholesterol storage disease, has highlighted important inflammatory, molecular and cellular pathways underlying its pathophysiology, leading to a better characterization of these patients. Despite these advances, CVDs still remain the leading cause of morbidity and mortality worldwide.7
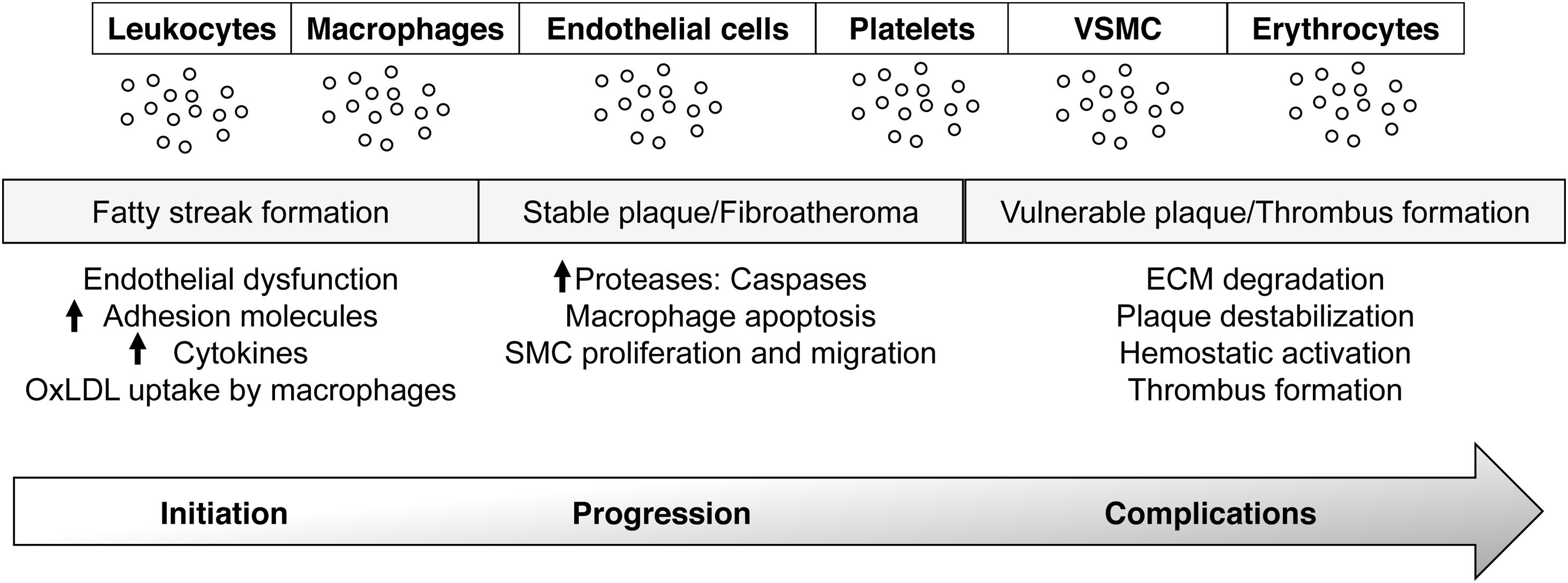
Pathophysiology of atherosclerosisThe vascular endothelium, as an integral component of the CV system, plays a crucial role in maintaining systemic homeostasis, by regulating vascular muscle contraction, relaxation, smooth muscle proliferation, and the expression of adhesion molecules or chemotactic factors.8 By producing vasoactive, anti-inflammatory, antithrombotic and cytostatic substances, the endothelium maintains the vascular tone, and prevents the adhesion of inflammatory cells and platelets, keeping at bay the coagulation and the fibrinolytic systems. This balance can be broken under conditions of oxidative stress, hyperlipidemia, hypertension, diabetes or smoking, which induce endothelial activation.9,10 The disruption of these haemostatic processes underpins the mechanism of atherosclerosis, activating prothrombotic and proinflammatory pathways, that result in lipid, protein and nucleic acid oxidation, and in the production of reactive oxygen species (ROS), chemokines, cytokines and adhesion molecules, that enable leucocyte and platelet adhesion, and thrombus formation.9,10 A characteristic feature of the initial stages of atherosclerosis is the local accumulation of monocytes and lymphocytes in the dysfunctional inner membrane of the vessel wall.11 Recruited monocytes differentiate into macrophages and transform into foam cells through the uptake of extracellularly modified lipoproteins, mainly oxidized low-density lipoproteins (ox-LDL). If the atherogenic stimulus continues, the accumulation of ox-LDLs in the subendothelial space, coupled with inflammatory cell infiltration and vascular smooth muscle cell (VSMC) migration and proliferation from the media to the intima, can lead to atherosclerotic lesion formation. All the evidences support an active crosstalk between the cellular components of the vessel wall and the circulation, that might be in part facilitated by the extracellular vesicles (EVs) released during the atherogenic process.12 As such, the study of EVs phenotype, content, and biological activity emerge as an opportunity to gain insight in the pathophysiology of CVDs (Fig. 1).13,14
, development (stable plaque/fibroatheroma), and progression and rupture (vulnerable plaque/thrombus formation). VSMC: smooth muscle cells.")
EVs in vascular inflammation and atherosclerosis. EVs are released by all cellular components of the vascular wall and blood and participate in all steps of atherosclerosis; initiation (fatty streak formation), development (stable plaque/fibroatheroma), and progression and rupture (vulnerable plaque/thrombus formation). VSMC: smooth muscle cells.
More than 70 years ago, Chargaff and West hypothesize that platelets might not be the only source of prothrombotic material, and demonstrated that blood samples depleted of platelets were able to induce clotting. In addition, high-speed centrifugated plasma presented reduced thrombin generation capacity, whereas the recovered pellet was able to induce clotting in vitro, suggesting the presence of prothrombotic acellular particles in that sedimented fraction.15 Despite the technical limitations at that time, they even anticipated a possible role of these subcellular components in the modulation of physiological and pathological processes.15 In 1955, Hougie reported that the coagulant activity of citrated plasma increased during storage of some hours.16 Further on, in the late 1960s, Wolf identified platelet derived phospholipids-rich procoagulant subcellular elements in both plasma and serum, and was able to resolve them by electron microscopy, observing particles between 20 and 50nm. He called this particle subfraction platelet dust.17 From that moment on, the interest in EVs has grown exponentially, from being considered mere cellular waste disposal systems, to be pinpointed as important mediators of intercellular communication processes among a bunch of target cells and tissues.12
EVs biology and functionIn spite of current technical developments for nanoscale particle analysis, EVs size and their scarce biological content represent a huge limitation for their characterization, and hamper a consensus on their nomenclature or classification. Initially, these particle subpopulations were classified according to their size and biogenesis into: exosomes with diameters between 30 and 150nm, generated by the inward budding of the endosomal membrane18; microparticles, microvesicles or ectosomes referring to vesicles with 100–1000nm of diameter, directly shed from the plasma membrane and polydisperse in size; and finally, apoptotic bodies, the biggest subpopulation (1–5μm of diameter) generated during the apoptotic process that can even contain cellular organelles.19–23 Due to the considerable overlap in size and composition between exosomes and microvesicles, and the lack of specific separation methods for each particle fraction, later recommendations and guidelines encourage the use of the general term EVs in the absence of accurate information about their subcellular origin.23,24
EVs are spherical lipid bilayers containing cytoplasmic material that are released to the blood stream and other biological fluids (e.g.: saliva, urine) by most cells and tissues under certain physiological and pathological circumstances. The number of EVs and their content varies according to the cellular activation status and the stimulus triggering their formation and release, increasing under stress conditions. Likewise, EVs have been postulated as small reservoirs of proteins, lipids, metabolites and nucleic acids regulated by specific pathophysiological processes.25 Interestingly, their cargo is protected by the lipid bilayer from the activity of endogenous DNases, RNases or proteinases, as well as from adverse physicochemical conditions related to sample processing, such as prolonged storage, multiple freeze/thaw cycles or extreme pHs.26 These features, together with their ability to hold markers of the parental cell, have postulated EVs as an attractive component of the liquid biopsy. In this line, the analysis of their cargo might be useful to understand the pathophysiological condition behind their generation, leading to the identification of novel diagnostic and prognostic biomarkers, and therapeutic targets.23,27
EVs biological activity in neighbour or distant cells is displayed by different mechanism related to their content and their lipidic nature.22 EVs rich in bioactive molecules in their surface will be able to interact directly with target proteins or extracellular matrix components changing their activity or composition.28 EVs can also activate signalling pathways in host cells through different mechanisms, including receptor–ligand interactions, transference of their cargo to the cytoplasm by fusion with the plasma membrane, or through endocytosis, phagocytosis or micropinocytosis, regulating among other processes, vascular homeostasis and atherosclerosis.13,29
Separation and characterization of extracellular vesiclesThe absence of specific methods for EVs separation requires a careful experimental design according to the downstream application and the biological sample of origin. In this section we will give a short overview of the most widely used methods for EVs isolation and characterization. For more detailed and extensive information readers can consults the latest update of the International Society of EVs23 and other recent reviews and articles.30–32
When working with blood samples, it is important to consider not only the analytical, but also the preanalytical conditions starting from blood withdrawal (e.g.: citrated vs EDTA plasma, gauge diameter, etc.), posterior blood processing (centrifugation speed, temperature, etc.) or plasma storage,33–35 since this will predetermine ulterior EVs separation technique, favouring yield against purity or vice versa based on the final purpose of the study.
Ultracentrifugation (UC) is the most widely used technique for EVs separation,32,36 but requires high amount of starting material and contaminating protein aggregates (e.g.: HDL, LDL, VLDL, or chylomicrons) are pelleted together with EVs.23,37 This can improve combined with density gradient (commonly with sucrose) that notably diminishes contaminants, but can compromise EVs yield. Moreover, either UC alone or UC+density gradient are time-consuming and low-throughput methods, limiting their applicability in clinical scenario. Size exclusion chromatography (SEC) is a faster isolation method (∼15min) that efficiently enriches and separates EVs from contaminants. Its yield can be increased combined with ultrafiltration, although abundant proteins as albumin might obstruct the nanopores, compromising both concentration and purity.38 Polymer-based precipitation methods, either hand-made or commercial, are effective for concentrating EVs but not for removing protein aggregates, and affinity-based techniques, using antibodies or proteins against specific EVs surface markers or receptors, are also commonly used for isolating particular EV subpopulations.39 Novel EVs separation methods including microfluidic-based devices, flow field-flow fractionation, or high-resolution flow cytometry are promising alternatives for EVs separation.40–43
Once separated, EVs should be characterized by different methods.23 Western blot is recommended to test the presence of specific EVs markers and common co-precipitated contaminants, nanoparticle tracking analysis (NTA) is useful to determine size and concentration of single particles,44 high-resolution flow cytometry with specific EV surface markers or labelled with nonfluorescent pro-dyes is used to test EVs markers and cellular origins,42,45 and conventional transmission electron microscopy (TEM) or cryo-TEM for EVs morphology and size assessment.46
These evidences highlight the need of a careful experimental design when EVs studies are performed, as well as a thorough description of the methods used for the separation and characterization to enable reproducibility.
Extracellular vesicles potential biomarkers in atherosclerosisCompelling evidences point towards an important role of EVs in all the stages of atherosclerosis and its associated complications.12,47 Indeed, all blood and vascular cells are capable of releasing EVs to the circulation, and elevated levels of platelet, endothelial, erythrocyte or leucocyte derived EVs have been associated with the presence of traditional CV risk factors, including diabetes,48,49 hypertension,50 hypercholesterolemia51,52 and smoking,53 as well as with subclinical and clinical atherosclerosis.54–57 In this regard, it has been reported a correlation of blood EVs and inflammation, and thrombotic risk in patients with coronary disease. Specifically, in coronary heart disease (CHD), endothelial and platelet derived EVs have been correlated with the inflammatory markers interleukin-6 (IL-6) and C-reactive protein (CRP),58 and displayed procoagulant activity in vitro.59 The levels of EVs have also been studied as possible markers of plaque instability. In fact, EVs of different cellular origins, e.g.: platelet, endothelial, leucocyte and erythrocyte derived, were increased in patients with myocardial infarction compared with patients with unstable and stable angina, and a relationship between procoagulant EVs, assessed by their content in tissue factor or annexin V, and CHD presentation was equally demonstrated.58–63 From a prognosis perspective, endothelial and erythrocyte EVs were associated with worse vascular function and CV events in coronary patients,64–69 and with poor outcome in stroke.70–72 Similarly, platelet and endothelial derived EVs have been found elevated in PAD and correlated with disease severity,73–76 although the role of EVs in PAD should be reconsidered according to their cargo. For instance, Crawford et al. found an elevation of monomeric CRP+ endothelial EVs in PAD, and suggested their contribution to subclinical inflammation,76 while Giarretta et al. proposed that Sonic hedgehog morphogen positive endothelial EVs could participate in angiogenic processes in skeletal muscle.77 In this regard, the transcriptomic study of EVs led us to identify calprotectin and lipocalin-2 as potential biomarkers of worse prognosis in PAD,55,78 and the proteomic analysis of circulating and arterial EVs in abdominal aortic aneurysm (AAA) revealed their enriched in proteins related to oxidative stress, inflammation and thrombosis, key processes underlaying AAA pathophysiology.79–81
Extracellular vesicles as active players in atherosclerosisBesides their possible utility as CV risk biomarkers, EVs emerge as active players in the processes leading to atherosclerosis, including endothelial dysfunction, inflammation, vascular remodelling or apoptosis.25,82 In fact, circulating EVs from patients with CHD and metabolic syndrome reduced ECs viability and proliferation,83 while increasing endothelial permeability and trans-endothelial migration,84 and promoted VSMC dedifferentiation in vitro.84 These data support a crosstalk between the cellular components of the aortic wall and the circulating EVs.
Studies with in vitro and in vivo models of atherosclerosis (Table 1) suggest that the activity of EVs in vascular remodelling, being beneficial or detrimental, depends not only on their cellular origin, but also on their cargo, which varies according to stimulus/stimuli triggering their release. For instance, endothelial EVs obtained after ox-LDL stimulation (ox-LDL-EC-EVs), induced endothelial death or pyroptosis in vitro in a mechanism dependent on the long non-coding (lnc)RNA HIF1A-AS2, that inhibited miR-455-5p enabling the expression of its target gene ESRRG and the activation of caspase-1 and NLRP3 (NLR family pyrin domain containing 3), and in vivo ox-LDL-EC-EVs increased lesion size and induced apoptosis in arterial ECs of Apoe−/− mice.85 In this line, other authors reported a switch towards a proinflammatory (M1) phenotype of macrophages upon ox-LDL-EC-EVs exposure in vitro, through the overexpression of miR-155, that was prevented when ox-LDL EC-EVs were enriched in the lncRNA Kruppel-like factor 2 (KLF2).86 Indeed, Apoe−/− mice receiving KLF2+-EC-EVs presented smaller arterial lesion size and proinflammatory (M1) macrophages, while increasing the number of M2 anti-inflammatory cells supporting the protective effect of this specific EC-EVs subpopulation.86 In this line, it has been reported a reduction in VSMCs migration and proliferation in vitro, and neointima formation in a model of carotid artery injury by EC-EVs in a mechanism dependent on low-density lipoprotein receptor-related protein 6 (LRP6) and miR-126-3p.87
Summary of studies investigating the biological activity of EVs from different origins in vitro and in vivo.
| EVs type and source | Study type | Biological effect | Ref. |
|---|---|---|---|
| EVs from CHD patients vs control EVs | In vitro | CHD-EVs reduced HUVECs viability and proliferation. | 83 |
| Plasma EVs from metabolic syndrome (MetS-EVs) vs non-MetS patients (nMETS-EVs) | In vitro | MetS-EVs increased HAECs permeability, and HASMC migration, proliferation and inflammation in a mechanism related to Rap1 activity. | 84 |
| Ox-LDL-EC-EVs from HUVECs | In vitro and in vivo | In vitro, ox-LDL-EC-EVs rich in the LncRNA HIF1A-AS2 induced apoptosis in HUVECs, inhibiting miR-455-5p, and upregulating ESRRD and caspase-1 activity.Ox-LDL-EC-EVs increased atherosclerotic lesion size and ECs apoptosis in Apoe−/− mice. | 85 |
| Ox-LDL-EC-EVs from HUVECs | In vitro and in vivo | Ox-LDL-EC-EVs induced M1 polarization on THP-1 monocytes depending on miR-155, that was prevented when EVs were enriched in KLF2.KLF2+-EC-EVs reduced lesion size and proinflammatory macrophages in Apoe−/− mice. | 86 |
| EC-EVs from HCAECs | In vitro and in vivo | EC-EVs reduced VSMC migration and proliferation by transferring miR-126 to target cells, reducing LRP6.In vivo, EC-EVs decreased neointima formation in a model of carotid artery injury in WT mice. | 87 |
| M-EVs and ox-LDL-M-EVs from differentiated THP-1 macrophages | In vitro and in vivo | ox-LDL-M-EVs poor in miR-199-5p induce HAECs apoptosis through the regulation of the SMARCA4/PODXL/NF-κB axis.M-EVs treated Apoe−/− mice presented reduced lesion size and blood inflammatory markers. | 88 |
| ox-LDL-M-EVs from THP-1 cells | In vitro | Ox-LDL-M-EVs induced HUVECs apoptosis in a mechanism mediated by the LncRNA GAS5. | 89 |
| M-EVs and ox-LDL-M-EVs from primary cultures or RAW264.7 cells | In vitro | Ox-LDL-M-EVs reduced macrophage migration in mechanism involving miR-146a and the reduction in IGF2BP1 and HuR. | 90 |
| ox-LDL-M-EVs from RAW264.7 macrophages | In vitro and in vivo | In vitro, miR-19b-3p enriched ox-LDL-M-EVs increased VSMCs proliferation and migration, and decreased JAZF1 expression.In vivo, ox-LDL-M-EVs aggravated atherosclerosis lesions in Apoe−/− mice. | 91 |
| M1-M-EVs from M1 polarized THP-1 cells | In vitro and in vivo | M1-M-EVs increased blood levels of cholesterol, oxidative stress and inflammatory markers in Apoe−/− mice, presenting bigger atherosclerotic plaques.miR-185-3p enriched M1-M-EVs aggravated this phenotype through Smad7 downregulation. | 92 |
| ox-LDL-M-EVs from THP-1 and RAW264.7 macrophages | In vitro | In vitro, ox-LDL-M-EVs rich in miR-503-5p and poor in smad7, smurf1, and smurf2 impaired angiogenesis, and activated apoptosis and inflammation in HCAECs and induced HCASMCs proliferation and migration. | 93 |
| miR-155+VSMC-EVs from HASMCs | In vitro and in vivo | In vivo, miR-155+VSMC-EVs from KLF5− transfected cells increased lesion size, and disrupted endothelial integrity in Apoe−/− mice.miR-155+VSMC-EVs decreased the endothelial expression of ZO-1. | 94 |
| VSMC-EVs from diabetic and non-diabetic HCASMCs | In vitro and in vivo | In vitro, diabetic-VSMC-EVs increased ECs expression of ICAM-1 and monocyte adhesion, and induced monocyte M1 polarization in a miR-221/222 dependent manner.In Apoe−/− mice, diabetic-VSMC-EVs increased plaque size. | 95 |
| RBC-EVs from RBCs of healthy volunteers | In vitro and in vivo | RBC-EVs upregulated the expression of HO-1 and ABCG1, and induced the polarization of macrophages towards M2 in vitro.In vivo, RBC-EVs decreased aortic lesions, and increased macrophage HO-1 expression in Apoe−/− mice | 96 |
| PLT-EVs from mouse platelets | In vitro and in vivo | PLT-EVs reduced circulating total cholesterol and triglycerides, and lesion size and inflammatory markers in arterial tissue of Apoe−/− mice, transferring miR-34c-5p to target cells, and decreasing PODXL. | 97 |
| PVAT-EVs from WT mice | In vitro | The pre-treatment of RAW264.7 macrophages with PVAT-EVs reduced ox-LDL uptake, and induced cholesterol efflux in a mechanism involving miR-382-5p. PVAT-EVs increased macrophage expression of the cholesterol efflux transporters ABCA1 and ABCG1, and the PPARγ, and reduced the levels of BMP4 and SR-A, that was reversed in presence of a miR-382-5p inhibitor. | 98 |
ABCA1: ATP binding cassette subfamily A member 1; ABCG1: ATP binding cassette transporter-1; BMP4: bone morphogenetic protein-4; ECs: endothelial cells; EVs: extracellular vesicles; GAS5: growth arrest specific 5; HO-1: heme oxygenase-1; HAECs: human aortic endothelial cells; HASMC: human aortic smooth muscle cells; HCAECs: human coronary artery endothelial cells; HCASMCs: human coronary artery vascular smooth muscle cells; HuR: human antigen R or ELAV-like RNA-binding protein 1; IGF2BP1: insulin-like growth factor 2 mRNA-binding protein 1; JAZF1: zinc finger gene 1; KLF2-5: Kruppel-like factor; MetS: metabolic syndrome; NF-κB: nuclear factor kappa B; LPR6: low-density lipoprotein receptor-related-protein 6; ox-LDL: oxidized low-density lipoprotein; PVAT: perivascular adipose tissue; PPARγ: peroxisome proliferator-activated receptor gamma; PLT: platelets; PODXL: podocalyxin like; RBC: red blood cells; SMARCA4: SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4; SR-A: macrophage scavenger receptor-A; Smurf: SMAD specific E3 ubiquitin protein ligase; ZO-1: zonula occludens-1.
This dual role has also been observed for macrophage derived EVs (M-EVs). As such, the administration of M-EVs to Apoe−/− mice reduced atherosclerotic plaque size and serum inflammatory markers compared with untreated mice, and with mice receiving ox-LDL-M-EVs, suggesting that ox-LDL exposure might avert the beneficial effect of M-EVs in atherosclerosis.88 These authors described a reduction of the antiapoptotic miR-199-5p in ox-LDL-M-EVs, that when replenished, reduced EC pyroptosis in vitrovia the regulation of the SMARCA4/PODXL/NF-κB axis,88 and in vivo reduced lesion size and serum inflammatory markers, as well as caspase-1 and IL-1β expression in aortic endothelial cells.88 The proapoptotic activity of ox-LDL-M-EVs on ECs in vitro was further corroborated by other group, identifying the lncRNA GAS5 as a possible mediator.89 Besides ECs, M-EVs can also interact with surrounding macrophages and VSMCs. The exposure of naïve macrophages to ox-LDL-M-EVs inhibited migration to a greater extent than control-M-EVs in a mechanism involving miR-146a, and the reduction of its target genes IGF2BP1 (insulin-like growth factor 2 mRNA-binding protein 1) and HuR (human antigen R or ELAV-like RNA-binding protein 1).90 Additionally, in vitro, miR-19b-3p enriched ox-LDL-M-EVs increased VSMCs proliferation and migration through the decrease of its downstream gene, zinc finger gene 1 (JAZF1), while in vivo, aggravated atherosclerosis lesions in Apoe−/− mice.91 A worsening of the atherosclerotic phenotype was also observed in Apoe−/− mice injected with M-EVs obtained after M1 differentiation (M1-M-EVs), that resulted in increased blood levels of cholesterol, oxidative stress and inflammatory markers, and locally in bigger atherosclerotic plaques. This phenotype was further aggravated when M1-M-EVs overexpressed miR-185-3p and induced the downregulation of its target gene Smad7.92 Similarly, ox-LDL-M-EVs rich in miR-503-5p and poor in smad7, smurf1, and smurf2 impaired angiogenesis, and activated apoptosis and inflammation in ECs in vitro, while inducing proliferation and migration in VSMCs.93
Experiment performed stimulating ECs with KLF5+VSMC-EVs or ox-LDL-VSMCs-EVs, resulted into the transfer of miR-155 to the endothelial monolayer, pointing towards a potential role of this miRNA in atherosclerosis.94 As such, the injection of miR-155+VSMC-EVs to Apoe−/− mice increased lesion size, and promoted the disruption of endothelial integrity by decreasing the endothelial expression of tight junction proteins, suggesting a potential role of KLF5+VSMC-EVs in endothelial injury.94 Similarly, VSMC-EVs derived from diabetic donors increased the endothelial expression of the proinflammatory molecule ICAM-1 (intercellular adhesion molecule) and monocyte adhesion compared to non-diabetic-VSMC-EVs, and promoted M1 polarization of human monocytes in a miR-221/222 dependent manner. Finally, the intravenous administration of diabetic-VSMC-EVs, resulted in a significant increase of atherosclerotic plaque size in Apoe−/− mice as compared with non-diabetic-VSMC-EVs.95 These results suggest a detrimental effect of activated VSMC derived EVs on endothelial function, and the potential utility of EVs for novel biomarker and therapeutic target discovery in atherosclerosis.
The contribution of EVs from other sources, e.g.: blood cells, or other organs, to the atherosclerotic phenotype has also been investigated. For instance, in vitro, the stimulation of macrophages with red blood cells derived EVs (RBC-EVs) resulted in the upregulation of the athero-protective genes heme oxygenase-1 (HO-1) and the ATP binding cassette transporter-1 (ABCG1), and the polarization of macrophages towards a reparative phenotype.96 Moreover, RBC-EVs-primed macrophages showed reduced Oil Red O staining after ox-LDL challenge, suggesting that RBC-EVs protect macrophages against foam cell formation and atherosclerosis. In vivo, Apoe−/− mice treated with RBC-EVs showed a decrease in total aortic lesions with a concomitant increase in macrophage HO-1 expression.96 Similarly, platelet derived EVs (PLT-EVs) were athero-protective by reducing circulating total cholesterol and triglycerides, and arterial lesion size and inflammatory markers in Apoe−/− mice, in a mechanism mediated by the transfer of miR-34c-5p to target cells and the decrease of PODXL (podocalyxin like).97 A possible crosstalk facilitated by EVs between perivascular adipose tissue (PVAT) and macrophages has also been reported.98 Indeed, the pre-treatment of murine macrophages with PVAT-EVs reduced ox-LDL uptake in vitro, and induced cholesterol efflux in a mechanism involving the athero-protective miR-382-5p. PVAT-EVs increased macrophage expression of the cholesterol efflux transporters ABCA1 and ABCG1, and the peroxisome proliferator-activated receptor gamma (PPARγ), and reduced the mRNA levels of bone morphogenetic protein-4 (BMP4) and macrophage scavenger receptor-A (SR-A), that was reversed in the presence of a miR-382-5p inhibitor.98 These evidences point towards a beneficial effect of EVs derived from cells of non-vascular origin in atherosclerosis, although this should be corroborated with broader studies.
ConclusionsOverall, EVs emerge as important mediators of cellular communication in atherogenesis, being detrimental or beneficial according to their cellular origin, the stimuli triggering their release, and their cargo. This dual capacity holds promise for the discovery of novel diagnosis and prognosis biomarkers, as well as for the identification of new therapeutic targets in CVDs by the study and characterization of EVs.
FundingThe Foundation for Applied Medical Research, Universidad de Navarra (Spain); the Ministry of Science and Innovation, Institute of Health Carlos III, co-funded by the European Fund for Economic and Regional Development (FEDER) [PI21/00622, Gobierno de Navarra 8-2021]. The 8-2021 project has received 50% co-financed aid from the European Regional Development Fund through the ERDF 2014-2020 Operational Program of Navarra. This research was supported by CIBER – Consorcio Centro de Investigación Biomédica en Red – Cardiovascular (CB16/11/00483 and CB16/11/00451), Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación.
Authors’ contributionsConceptualization: CR; writing: JAP and CR; review and editing: JAP, AC, FC, and CR. All authors have read and agreed to the published version of the manuscript.
Conflict of interestsNone to declare.