




Apolipoprotein C-III (apoC-III) is a small protein that is predominantly synthesized in the liver and mainly resides at the surface of triglyceride-rich lipoproteins. Its expression is upregulated by glucose and reduced by insulin, with enhanced apoC-III promoting hypertriglyceridemia and inflammation in vascular cells. The protein is also elevated in patients with diabetes, suggesting that enhanced apoC-III levels might contribute to the development of type 2 diabetes mellitus. The present review focuses on the key mechanisms by which apoC-III could promote type 2 diabetes mellitus, including exacerbation of insulin resistance in skeletal muscle, activation of β-cell apoptosis, promotion of weight gain through its effects on white adipose tissue and hypothalamus, and attenuation of the beneficial effects of high-density lipoproteins on glucose metabolism. Therapeutic strategies aimed at reducing apoC-III levels may not only reduce hypertriglyceridemia but also might improve insulin resistance, thus delaying the development of type 2 diabetes mellitus.
La apolipoproteína C-III (apoC-III) es una pequeña proteína predominantemente sintetizada en el hígado y que se encuentra principalmente en la superficie de las lipoproteínas ricas en triglicéridos. Su expresión es aumentada por la glucosa y reducida por la insulina, y sus niveles elevados promueven la hipertrigliceridemia, así como la inflamación en células vasculares. Esta proteína también se encuentra elevada en los pacientes diabéticos, lo que sugiere que el aumento de esta apoproteína podría contribuir al desarrollo de la diabetes mellitus de tipo 2. Esta revisión aborda los mecanismos clave por los que la apoC-III podría promover la diabetes mellitus tipo 2, entre los que se encuentran la exacerbación de la resistencia a la insulina en el músculo esquelético, la activación de la apoptosis en la célula β, la promoción del aumento de peso por sus efectos sobre el tejido adiposo blanco y el hipotálamo, y la atenuación de los efectos beneficiosos de las lipoproteínas de alta densidad sobre el metabolismo de la glucosa. Las estrategias terapéuticas dirigidas a disminuir los niveles de apoC-III no sólo podrían reducir la hipertrigliceridemia, sino también mejorar la resistencia a la insulina y retrasar el desarrollo de la diabetes mellitus de tipo 2.
Common to insulin resistant states in patients with obesity and type 2 diabetes mellitus (DM) is the presence of atherogenic dyslipidemia, which is known to amplify cardiovascular risk.1,2 Atherogenic dyslipidemia is characterized by elevated triglyceride levels, reduced high-density lipoprotein (HDL) cholesterol levels, and the appearance of small dense low-density lipoprotein (LDL).1,2 Interestingly, atherogenic dyslipidemia usually precedes a diagnosis of type 2 DM by several years, suggesting that lipoprotein abnormalities are an early event in the disease's development.3 In insulin resistant states, atherogenic dyslipidemia is initiated by an overproduction of triglyceride-enriched very low-density lipoproteins (VLDLs) that triggers the observed sequence of lipoprotein changes.1,2
In addition to triglycerides, VLDLs also contain apolipoproteins, of which apolipoprotein C-III (apoC-III) is one of the most abundant.4 It is well-known that the increase in apoC-III contributes to elevated triglyceride levels5,6 and cardiovascular risk.7,8 In fact, individuals with loss-of-function mutations in the APOC3 gene present lower triglyceride levels and a reduced risk of cardiovascular disease.9,10 ApoC-III promotes atherosclerotic plaque formation not only through its indirect effects on triglyceride levels but also through its direct pro-inflammatory effects on vascular cells.11 However, the effects of apoC-III on insulin resistance are less well-known.
Plasma apoC-III levels are elevated in patients with DM,4 which in turn, are associated with insulin resistance.12 This suggests that the increase in apoC-III during the development of atherogenic dyslipidemia might exacerbate insulin resistance, thereby accelerating the development of type 2 DM. Consistent with this, mice overexpressing apoC-III have been shown to be more prone to insulin resistance and DM induced by high-fat diet (HFD).13,14
In this manuscript, we aim to review the possible mechanisms that lead to apoC-III-induced insulin resistance.
ApoC-III: features and regulationApoC-III, which is encoded by the gene APOC3, is a major component of triglyceride-rich lipoproteins (TRL) (chylomicrons and VLDL) that is also present in LDL and HDL. Predominantly synthesized in the liver, and to a lesser extent in the intestine,15 its hepatic expression is negatively regulated by insulin through forkhead box protein O1 (FOXO1).16 This transcription factor binds to a consensus site in the APOC3 promoter and upregulates its expression. Insulin then phosphorylates and prevents FOXO1 translocation to the nucleus, thereby downregulating apoC-III expression. In animal models of insulin deficiency or resistance, a reduction in the effects of this hormone leads to unrestrained apoC-III production that provides potential mechanism for increased apoC-III levels, hypertriglyceridemia, and atherogenic dyslipidemia in diabetic patients. By contrast, glucose upregulates apoC-III expression in primary rat hepatocytes and in immortalized human hepatocytes via the transcription factors carbohydrate response element-binding protein (ChREBP) and hepatocyte nuclear factor-4α.17 This regulation of apoC-III by glucose may help to explain the link between hyperglycemia, hypertriglyceridemia, and cardiovascular disease in patients with type 2 DM. In addition, because peroxisome proliferator-activated receptor (PPAR)α activation by fibrates reduces the expression of APOC3 by decreasing hepatocyte nuclear factor-4α levels and promoting the displacement of this transcription factor from the APOC3 promoter,18 this mechanism provides an explanation for the reduction in plasma apoC-III levels with fibrate treatment. Farnesoid X receptor agonists also reduce hepatic APOC3 expression, an effect that may contribute to the hypotriglyceridemic effect of these compounds.19 Finally, the increase in plasma saturated fatty acids levels might result in hypertriglyceridemia by upregulating apoC-III expression via the PPARγ coactivator-1β (PGC-1β).20
ApoC-III exists as three isoforms that differ in terms of glycosylation: apoC-III0 (no sialic acid bound to the protein), apoC-III1 (one sialic acid residue), and apoC-III2 (two sialic acid residues). Each isoform contributes approximately 10, 55 and 35%, respectively, of the total circulating apoC-III levels.21 ApoC-III glycosylation seems to be under metabolic control since a reduction in the apoC-III1 to apoC-III2 ratio has been reported after weight loss by caloric restriction,22 whereas an increase in apoC-III0 has been observed in combined hyperlipidemia.23
ApoC-III in cardiovascular diseaseApoC-III is considered an independent risk factor for cardiovascular disease because of its close association with hypertriglyceridemia and inflammation in vascular cells.6 The effects of apoC-III on triglyceride levels can be explained by several mechanisms. First, it is a potent inhibitor of lipoprotein lipase (LPL), a key enzyme for the lipolysis of triglyceride from VLDL and chylomicrons.24 Second, it favors hypertriglyceridemia by attenuating the apoE-mediated hepatic uptake of TRL remnants by LDL receptors and LDL receptor-related protein 1.25–27 This indicates that apoC-III modulates triglyceride levels through LPL-dependent and LPL-independent mechanisms. More recent studies have shown that lowering apoC-III levels in the absence of apoE did not improve TRL clearance, but that it significantly decreased serum triglyceride levels.28 The hypotriglyceridemic effect in the absence of apoE resulted from improved LPL activity in white adipose tissue, rather than in all LPL-target tissues. Moreover, apoC-III stimulates the assembly and secretion of VLDL.29,30 Finally, in vitro studies using apoC-III complexes with higher apoC-III2/apoC-III1 ratios show attenuated inhibition of VLDL uptake by hepatic cells and LPL-mediated lipolysis, providing possible functional explanations for the inverse association between a higher apoC-III2/apoC-III1 ratio and hypertriglyceridemia and cardiovascular risk.31
The above mechanisms mean that elevated levels of apoC-III result in hypertriglyceridemia. Consistent with this, volanesorsen, a second-generation antisense oligonucleotide that inhibits apoC-III synthesis by coupling to the APOC3 messenger RNA, reduces serum triglycerides levels by 68% and increases HDL cholesterol levels by 40% in patients with DM, familial chylomicronemia syndrome, or with moderately high triglyceride levels.32 Genomic studies have also demonstrated the influence of apoC-III on cardiovascular risk. The Sstl polymorphism, that results in increased apoC-III levels, carries a higher risk of hypertriglyceridemia and cardiovascular disease.33 By contrast, loss-of-function mutations in the APOC3 gene have been shown to reduce non-fasting triglyceride levels by 44% and the incidence of ischemic heart disease by 36%.9
Regarding inflammation in vascular cells, apoC-III-rich lipoproteins or apoC-III itself can activate monocytes and stimulate their adhesion to vascular endothelial cells, thereby promoting atherosclerosis.7,8 Moreover, apoC-III upregulates the expression of vascular cell adhesion molecule-1 in endothelial cells to increase monocyte adhesion.7 These cells later differentiate into tissue macrophages, leading to inflammation, transformation in foam cells, and atherogenesis. Notably, the stimulation of inflammatory pathways can exacerbate the increase in apoC-III levels because activation of the pro-inflammatory transcription factor nuclear factor κB (NF-κB) upregulates the expression of apoC-III.34
ApoC-III can also affect a critical step in the pathogenesis of atherosclerosis, the deposition and retention of lipoproteins by vascular extracellular matrix molecules, particularly proteoglycans.35 Although, apoC-III does not bind directly to proteoglycans, it has been reported that increases in apoC-III content alters the lipid composition in LDL from diabetic patients with a high endogenous apoC-III/apoB molar ratio, which allows apoB to acquire a conformation that is more favorable for proteoglycan binding.11
The marked heterogenicity in apolipoprotein content in HDLs affects their function. ApoC-III is present in 4% of HDL in subjects of normal weight, but it increases to 10% in subjects who are obese.36 This may be responsible for attenuating some of the beneficial effects of HDL, because it is known that there is a higher risk of cardiovascular disease than when compared to HDL that lacks apoC-III.37 Similarly, HDLs from patients with coronary artery disease have been shown to stimulate endothelial proapoptotic pathways because of the higher content of apoC-III; by contrast, HDL with less apoC-III has been shown to inhibit apoptosis.38 These findings indicate that HDL containing apoC-III may be dysfunctional.
ApoC-III and insulin resistanceInsulin resistance and β-cell failure are the two major pathophysiologic abnormalities that contribute to the development of type 2 DM. Insulin resistance precedes and predicts the development of type 2 DM and is initiated when overnutrition results in elevated levels of glucose and/or lipids in the serum, inducing low-grade inflammation and activating various transcriptional and metabolic pathways. This results in the induction of various pro-inflammatory mediators that provoke the pathogenesis of tissue-specific insulin resistance by attenuating insulin signaling pathways.39 Once insulin resistance develops, it promotes β-cell failure, and impaired insulin secretion, ultimately resulting in overt hyperglycemia.
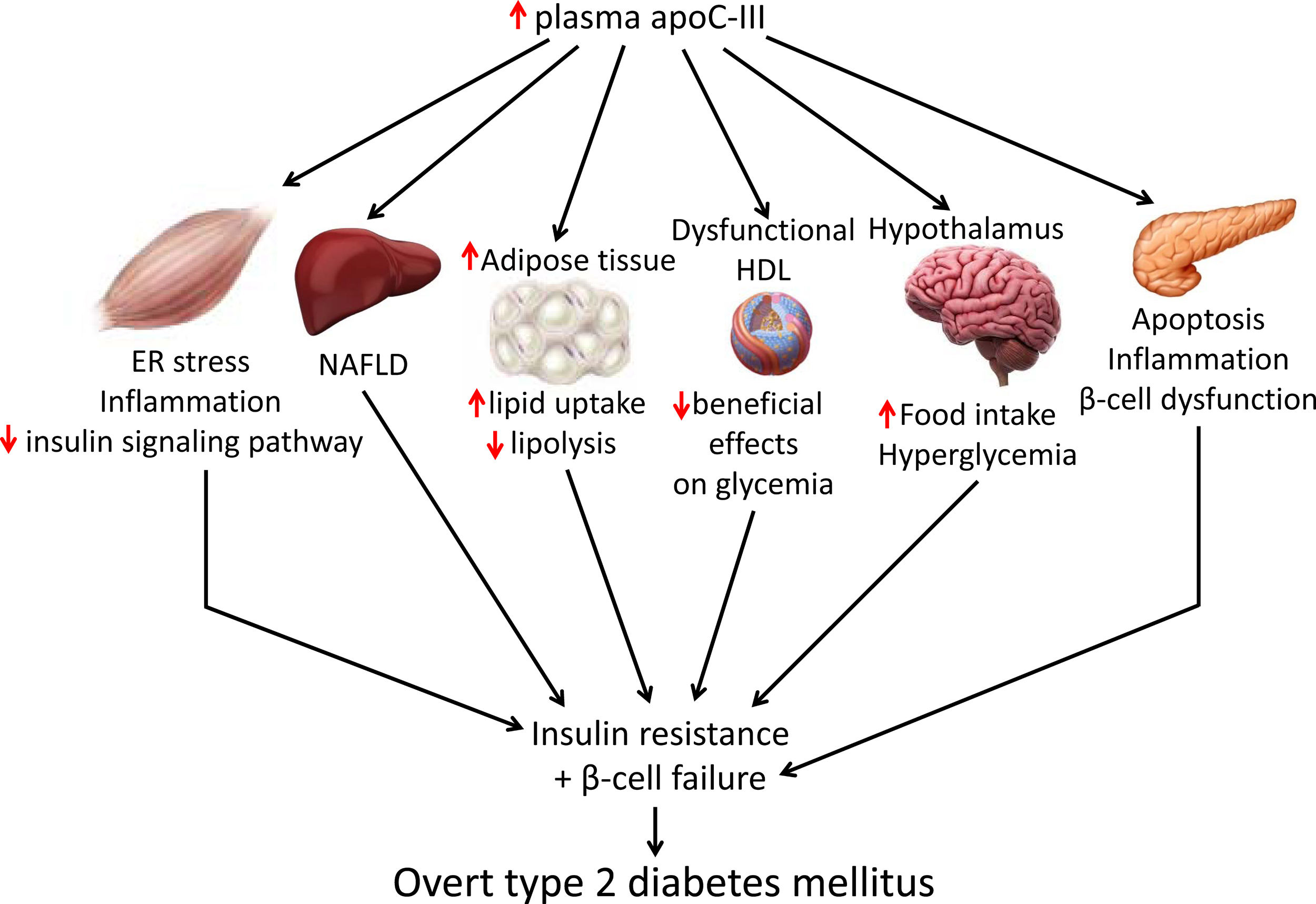
Skeletal muscle insulin resistance is the primary defect that precedes β-cell failure and overt hyperglycemia by decades.40 In fact, the primary site of insulin-stimulated glucose disposal is skeletal muscle, accounting for up to 90% of glucose clearance.41 Consequently, loss of skeletal muscle insulin sensitivity is critical to the pathogenesis of type 2 DM.42 Although the mechanisms involved in the development of insulin resistance are currently unclear, accumulating evidence points to the presence of a chronic low-level inflammatory process.39 Endoplasmic reticulum (ER) stress43 and toll-like receptors (TLRs)44 have been implicated in the activation of pro-inflammatory kinases, such as the inhibitor of κB (IκB) kinase β (IKK-β) in the NF-κB pathway. Together with other kinases, this phosphorylates the insulin receptor substrate 1 (IRS-1) in serine residues and attenuate the insulin signaling pathway. Likewise, pro-inflammatory pathways upregulate the expression of multiple inflammatory mediators that also facilitate insulin resistance.39 We have previously reported that skeletal muscle from transgenic mice overexpressing apoC-III show increased levels of ER stress and inflammatory markers (Fig. 1).45 Activation of ER stress by apoC-III in myotubes stimulated the NF-κB pathway, attenuated insulin signaling by increasing the phosphorylation of IRS-1 in serine residues, and upregulated the expression levels of inflammatory mediators, including interleukin 6, tumor necrosis factor α, and monocyte chemoattractant protein-1. Remarkably, these effects were prevented by incubation with a neutralizing antibody against TLR-2, indicating that some of the effects of apoC-III might be mediated by this receptor. Given that apoC-III is the most abundant apolipoprotein in the VLDL of people with DM,4 these results suggest that their elevated VLDL may affect skeletal muscle and thereby exacerbate insulin resistance.

Non-alcoholic fatty liver disease (NAFLD) is the principal manifestation of liver disease in insulin resistant states such as obesity and metabolic syndrome. There are conflicting results for the effects of apoC-III in NAFLD. Transgenic mice overexpressing apoC-III fed a standard diet showed NAFLD-like features compared with non-transgenic control littermates, with these changes being exacerbated by feeding a HFD.46 Treatment with the PPARα activator fenofibrate reversed several of the effects caused by elevated apoC-III levels but failed to normalize inflammatory markers even when the increase in liver lipid accumulation was completely abolished.44 However, in a study where transgenic mice overexpressing apoC-III were fed a HFD for up to 10 months, the authors concluded that apoC-III was not a predisposing factor for linking overnutrition to NAFLD in obesity.47 In humans, carriers of APOC3 variant alleles develop increased plasma levels of apoC-III that is associated with NAFLD and insulin resistance.12,48 By contrast, several studies show no causal relationship between APOC3 variants linked to hypertriglyceridemia and the hepatic triglyceride content49 or linking APOC3 promoter region polymorphisms to liver damage (severity of steatosis, non-alcoholic steatohepatitis, and moderate/severe fibrosis).50
Transgenic mice overexpressing apoC-III have also been used to confirm the role of this apolipoprotein in obesity. Diet-induced obesity was exacerbated in these mice due to the increased availability of free fatty acids from post-prandial TRLs, the greater adipose capacity for lipid uptake, and the reduced lipolysis in adipose tissue.51
ApoC-III also links insulin resistance and β-cell failure in type 2 DM. Local insulin resistance in β-cells reduces FOXO1 nuclear exclusion and increases apoC-III levels. The increase in islet apoC-III then causes apoptosis by increasing cytoplasmic free Ca2+ concentrations and promoting a local inflammatory milieu.52 Supporting this, antisense oligonucleotide treatment decreased in vivo apoC-III and improved glucose tolerance. ApoC-III knockout islets transplanted into diabetic mice with high systemic levels of apoC-III have also been shown to induce normal cytoplasmic free Ca2+ concentrations in the absence of inflammation. In the setting of islet insulin resistance, these findings indicate that locally produced apoC-III impairs β-cell function and contributes to the development of type 2 DM. In type 1 diabetic patients, increased activity of voltage-gated L-type Ca2+-channels in β-cells results in increased cytoplasmic free Ca2+, leading to β-cell apoptosis. Remarkably, it has been reported that in these patients apoC-III stimulates the activity of L-type voltage-gated Ca2+-channels in insulin-producing cells, ultimately leading to an increase in cytoplasmic free Ca2+ and β-cell apoptosis.53
Research indicates that HDL ameliorates glucose metabolism by modulating insulin secretion by pancreatic β-cells,54–56 and insulin-independent glucose uptake by skeletal muscle,56,57 as well as by inhibiting inflammation.58 Although results conflict, the presence of apoC-III in HDL may attenuate the beneficial effects of these lipoproteins on glucose metabolism. Thus, dysfunctional HDL containing apoC-III was reported to be a major independent predictor of new-onset type 2 DM.59 A more recent study confirmed that the presence of apoC-III on HDL also diminished the protective association of HDL with incident DM.60 In another study, however, HDL without apoC-III was inversely associated with risk of DM, but no such association was shown for HDL with apoC-III.61
ApoC-III might also affect food intake by inhibiting LPL in the hypothalamus. This lipase promotes fatty acid uptake in this brain tissue, ultimately downregulating orexigenic neuropeptide expression, which in turn, reduces food intake and inhibits glucose production.62,63 ApoC-III is expressed in the hypothalamus, and its intracerebroventricular injection can suppress hypothalamic LPL activity and stimulate night-time food intake.64 These findings suggest that increased apoC-III levels in the hypothalamus can promote food intake, leading to obesity and altered glucose metabolism.
Finally, genetic and pharmacological approaches have provided robust evidence of the involvement of apoC-III in the development of insulin resistance. Data show that subjects with a genetic mutation in the APOC3 gene, who have life-long reduced apoC-III levels, are healthier overall and have increased insulin sensitivity.65 Moreover, a small study showed that volanesorsen improved dyslipidemia and insulin sensitivity when used to treat patients with type 2 DM.66 Although the cohort was small, a strong relationship was found between enhanced insulin sensitivity and both plasma apoC-III and triglyceride suppression.
Targeting apoC-IIIApoC-III has a key role in lipid metabolism and inflammation, but it can also contribute to the development of type 2 DM by promoting insulin resistance in skeletal muscle and triggering β-cell apoptosis. It also contributes to obesity through effects on white adipose tissue and hypothalamus and attenuates the beneficial effects of HDL on glucose metabolism. Therefore, lowering apoC-III reduces not only cardiovascular risk but also insulin resistance, which in turn is involved in the development of atherogenic dyslipidemia and serves as a risk factor for type 2 DM and cardiovascular disease. Several therapeutic tools are available for reducing apoC-III, including the PPARα activators fibrates, which reduce hepatic apoC-III expression,19 or the insulin sensitizer pioglitazone, a PPARγ activator used in the treatment of type 2 DM.67 A meta-analysis of randomized clinical trials also reported that statins can decrease apoC-III levels, helping to explain their hypotriglyceridemic action.68 However, directly targeting APOC3 mRNA with volanesorsen is the only treatment that induces a robust reduction in apoC-III levels.
The clinical efficacy and safety of volanesorsen as a lipid-lowering drug has recently been reported in phase 2 and phase 3 clinical studies.32 It induced a strong reduction in serum triglyceride levels and a significant increase in HDL cholesterol. However, larger clinical trials are needed to validate the use of volanesorsen as a lipid-lowering drug for eliminating residual cardiovascular risk. Concerning safety issues, the APPROACH trial in patients with familial chylomicronemia syndrome revealed that volanesorsen caused thrombocytopenia in 76% of treated patients.69 However, no significant differences have been reported between carriers and non-carriers of an inactivating mutation in APOC3 in platelet count or in the prevalence of thrombocytopenia.70 This suggests that this side effect is drug- or class-specific rather than a consequence of apoC-III inhibition.
A monoclonal antibody targeting lipoprotein-bound human apoC-III has also been developed.71 In mice expressing human APOC3, its administration promoted circulating apoC-III clearance and enhanced TRL catabolism. To date, its effects on insulin sensitivity has not been evaluated.
ConclusionsThe available evidence suggests that apoC-III is not only relevant to lipoprotein metabolism and inflammation but also to insulin resistance, with evidence that pharmacological targeting its reduction might improve this latter condition. Drugs reducing apoC-III levels or activity might be key in type 2 diabetic patients, since cardiovascular disease remains the principal cause of death among these patients. Additional research is needed to confirm the efficacy of volanesorsen in patients with type 2 DM because only a single study with a small number of patients has examined this potential new indication. Moreover, safety concerns remain given the effects of volanesorsen on the platelet count and thrombocytopenia.
Authors’ contributionsAll authors contributing to the writing, editing, and approval of the manuscript.
Financial supportThis work was funded by the SEA-FEA grant for basic reseach 2017 and the Spanish Ministry of the Economy and Competitiveness (SAF2015-64146-R and RTI2018-093999-B-100 to MVC) and European Union ERDF funds. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is a Carlos III Health Institute projects.
This study was supported by the SEA-FEA grant for basic research 2017.
Conflict of interestThe authors declare no competing interests.