




Abdominal aortic aneurysm (AAA) is a multifactorial, degenerative disease characterized by progressive aortic dilation and chronic activation of inflammation, proteolytic activity, and oxidative stress in the aortic wall. The immune response triggered by antibodies against antigens present in the vascular wall participates in the formation and progression of AAA through mechanisms not completely understood. This work analyses the function of specific IgG receptors (FcγR), especially those expressed by monocytes/macrophages, in the development of experimental AAA.
MethodsIn the elastase-induced AAA model, the abdominal aortas from wildtype and FcγR deficient mice with/without macrophage adoptive transfer were analysed by histology and quantitative PCR. In vitro, RAW 264.7 macrophages were transfected with RNA interference of FcγRIV/CD16.2 or treated with Syk kinase inhibitor before stimulation with IgG immune complexes.
ResultsMacrophage adoptive transfer in FcγR deficient mice increased the susceptibility to AAA development. Mice receiving macrophages with functional FcγR exhibited higher aortic diameter increase, higher content of macrophages and B lymphocytes, and upregulated expression of chemokine CCL2, cytokines (TNFα and IL-17), metalloproteinase MMP2, prooxidant enzyme NADPH oxidase-2, and the isoforms FcγRIII/CD16 and FcγRIV/CD16.2. In vitro, both FcγRIV/CD16.2 gene silencing and Syk inhibition reduced cytokines and reactive oxygen species production induced by IC in macrophages.
ConclusionsActivation of macrophage FcγR contributes to AAA development by inducing mediators of inflammation, proteolysis, and oxidative stress. Modulation of FcγR or effector molecules may represent a potential target for AAA treatment.
El aneurisma aórtico abdominal (AAA) es una patología degenerativa y multifactorial caracterizada por una dilatación progresiva de la aorta y activación crónica de inflamación, actividad proteolítica y estrés oxidativo en la pared vascular. La respuesta inmune dependiente de anticuerpos IgG frente a antígenos expuestos en el vaso dañado está implicada en la formación y progresión del AAA, aunque los mecanismos no son del todo conocidos. En este trabajo analizamos la funcionalidad de los receptores Fc de IgG (FcγR), en particular los expresados por el monocito/macrófago, en el desarrollo del AAA experimental.
MétodosEn el modelo de AAA inducido por perfusión aórtica de elastasa se examinaron, mediante histología y PCR cuantitativa, las aortas abdominales de ratones de fenotipo salvaje y de ratones deficientes en FcγR, sin y con transferencia adoptiva de macrófagos. In vitro, macrófagos murinos se transfectaron con RNA de interferencia de FcγRIV/CD16.2 o se trataron con un inhibidor de la quinasa Syk antes de la estimulación con inmunocomplejos (IC) de IgG.
ResultadosLa transferencia adoptiva de macrófagos a ratones deficientes en FcγR incrementó su susceptibilidad al desarrollo de AAA. En los ratones que recibieron macrófagos con FcγR funcionales se observó un mayor incremento del diámetro aórtico y del contenido de macrófagos y linfocitos B, así como un aumento en la expresión de la quimioquina CCL2, las citoquinas TNFα e IL-17, la metaloproteinasa MMP2, la enzima prooxidante NADPH oxidasa-2 y las isoformas FcγRIII/CD16 y FcγRIV/CD16.2. In vitro, tanto el silenciamiento génico de FcγRIV/CD16.2 como la inhibición de Syk en macrófagos redujeron la producción de citoquinas y anión superóxido inducida por IC.
ConclusionesLa activación de los FcγR en macrófagos contribuye al desarrollo de AAA a través de la inducción de mediadores de inflamación, proteólisis y estrés oxidativo. La modulación de FcγR o sus moléculas efectoras podría ser una potencial diana para el tratamiento del AAA.
Abdominal aortic aneurysm (AAA) is a degenerative process of the vascular wall in which progressive dilatation of the aorta occurs, generally in its infrarenal portion, and is considered pathological when the diameter of the vessel is greater than or equal to 3 cm. AAAs are mostly asymptomatic and are often diagnosed by chance through imaging tests. Smoking, advanced age, male sex, hypertension, and hypercholesterolaemia are the main risk factors for AAA, which has a prevalence of 1%–2% in men over 65 years of age. The main complication of AAA is aortic rupture, which causes acute haemorrhage and a high mortality rate. There is currently no pharmacological treatment that can prevent or limit the progression of AAA. In symptomatic cases, or when the aneurysm diameter exceeds 5–5.5 cm, open or endovascular repair surgery is performed to mitigate the risk of rupture.1,2
The pathophysiology of AAA is complex, multifactorial, and not fully understood. Pathological features include progressive degradation of elastic fibres, inflammatory infiltration, vascular smooth muscle cell (VSMC) dysfunction, and increased oxidative stress.3 AAA is now considered a chronic inflammatory process involving the innate and adaptive immune system.4 It is known that during vascular damage there is a gradual infiltration of neutrophils, macrophages, T lymphocytes, mast cells, and NK cells, whose activation releases inflammatory mediators and reactive oxygen species (ROS) and promotes matrix metalloproteinase (MMP) activity and VSMC apoptosis, leading to weakening of the vascular wall and rupture of the AAA. Antibody-producing B cells have also been detected in AAA lesions and different antibodies, predominantly of the IgG isotype, have been identified that are able to recognise circulating proteins or components of the vascular wall and activate the inflammatory response in the aneurysmal tissue, although the effector molecules of this autoimmune response are not fully understood.6,7
IgG and immune complexes (IC) formed by antigen-IgG binding are able to interact with immune and tissue cells through receptors specific for the Fc (constant) portion of IgG (FcγR), activating a wide range of biological responses such as phagocytosis, antibody-dependent cytotoxicity, and release of inflammatory mediators. Functionally, these receptors are classified into activators (in humans; FcγRI/CD64, FcγRIIA,C/CD32A,C, and FcγRIIIA,B/CD16A,B; in mice; FcγRI/CD64, FcγRIII/CD16, and FcγRIV/CD16.2), and the inhibitor FcγRIIB/CD32B (in mice and humans), which differ in specificity, affinity, cellular distribution, and signalling pathways. Several activating isoforms associate intracellularly with the γ-chain, the polypeptide containing the ITAM activation motif involved in signal transduction through activation of Src and Syk kinases.8 Studies in patients and experimental models have shown that the balance between activator/inhibitor isoforms of FcγR and its ligands regulates various processes involved in the development of cardiovascular diseases, including endothelial function, thrombosis, hypertension, atherosclerosis, and inflammation.9–11 In the context of AAA, our group has recently described that deficiency in the γ-chain common to several FcγR activators limits the development of lesions in mice.12 In the present work we analyse, by means of in vivo adoptive transfer studies and in vitro experiments, the potential contribution of macrophage FcγR to the inflammatory response, proteolytic activity, and oxidative stress during experimental AAA formation.
Materials and methodsExperimental model of AAAThe care and maintenance of the animals, as well as the procedures carried out in this study, were performed under the 3 Rs principle (replacement, refinement, reduction), in accordance with European regulations (Directive 2010/63/EU), and were approved by the Animal Welfare Ethical Committee of the institution and the autonomous community (PROEX 116/16 and 217/19). γ-chain deficient mice (GKO) and wildtype (WT) controls, both of the C57BL/6J strain (Jackson Lab), were used. The mice were housed in ventilated cages (2–4 mice per cage) with customary bedding material and environmental enrichment in a conventional room with controlled temperature (20 °C–22 °C), 12 h light/dark cycle, and free access to standard water and chow. AAA induction was performed in 10−12-week-old male mice by intra-aortic perfusion of porcine pancreatic elastase type I (specific activity 7 U/mg; E1250; Sigma-Aldrich).12 Briefly, the mice were anaesthetised by inhalation of 2% isoflurane and a horizontal laparotomy was performed. Using a surgical stereomicroscope, the abdominal aorta was separated from the left renal vein up to the bifurcation and temporarily ligated between the renal and iliac arteries. An aortotomy was performed, a PE-26 polyethylene tube was inserted, and the aorta was perfused with elastase for 5 min at 100 mmHg. The aortotomy was then repaired, the ligation was removed, and restoration of blood flow was visually confirmed. The incisions were closed, and the mice were housed under standard conditions. For adoptive macrophage transfer, bone marrow cells were harvested from femurs of WT donor mice and differentiated to macrophages by incubation in DMEM medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 10% foetal bovine serum (FBS), and 10% L-929 cell-conditioned medium. After 7 days, macrophages were detached from the culture plates and 2 × 106 cells were injected intravenously into the GKO recipient mice, starting the day before elastase perfusion and every 4 days for the next 14 days. On day 14 after surgery, the mice were anaesthetised (100 mg/kg ketamine and 15 mg/kg xylazine), perfused with saline through the left ventricle, euthanised, and the aorta removed. The infrarenal region of the abdominal aorta was paraffin-embedded for histology studies (n = 5 mice per group) or processed for gene expression analysis (n = 4 mice per group).
Histological analysisSerial cross-sections of 4 μm of the abdominal aorta (over approximately 200 μm) were deparaffinised and stained alternately with Masson’s trichrome. For each mouse, aortic diameter was measured at the peak of the lesion, with AAA defined as a ≥100% increase in diameter. For immunohistochemical analysis, samples were deparaffinised and rehydrated, antigenic recovery was performed with citrate buffer and endogenous peroxidase was inactivated with 3% H2O2:methanol (1:1). After blocking non-specific binding (PBS with 4% albumin and 8% host serum of the secondary antibody), samples were incubated for 16 h at 4 °C with primary antibody against macrophages (CD68, Abcam Cat# ab53444, RRID:AB_869007) and B lymphocytes (CD45R, BD Biosciences Cat# 550286, RRID:AB_393581). After washing, the sections were incubated with biotinylated secondary antibody, followed by avidin-biotin-peroxidase complex and its chromogen (diaminobenzidine or aminoethylcarbazole), and counterstained with haematoxylin. Quantification of positive staining was performed blinded in at least 2 sections per mouse using Image-Pro Plus software and expressed as percentage of positive area with respect to the total lesion area.
Cell culture, inhibition, and silencingThe RAW 264.7 (ATCC-TIB-71) murine macrophage cell line was maintained in DMEM medium supplemented with antibiotics, L-glutamine, and 10% de FBS. The cells were maintained in medium without FBS for 16 h, pretreated for 1 h with Syk inhibitor (Bay 613606, 500 nM), and stimulated for 6 and 24 h with 150 μg/mL of soluble IgG aggregates. IgG aggregates were obtained by heating murine IgG (10 mg/mL, 30 min at 63 °C) and subsequent centrifugation. For gene silencing, the cells were transfected in Opti-MEM medium with 20 nmol small interfering RNA (siRNA) specific for FcγRIV/CD16.2 (siCD16.2; sc-42759, Santa Cruz Biotech) or with a non-specific siRNA as a negative control (siScr, 4390843, Thermo Fisher) using Lipofectamine RNAiMAX reagent (13778075, Thermo Fisher). Silencing was verified by Western blotting with anti-FcγRIV/CD16.2 antibody (Santa Cruz Biotech) and α-tubulin (Sigma-Aldrich) as loading control.
mRNA expressionTotal RNA from aorta and cells was extracted with TRI reagent (Molecular Research Centre). From 1 μg of RNA, cDNA was obtained by reverse transcription reaction using the High Capacity cDNA Archive kit (Applied Biosystems) in a conventional thermal cycler (PTC-100, MJ Research Inc.) under the following conditions: 10 min at 25 °C, 2 h at 37 °C, and 5 min at 85 °C. Expression levels of target genes were determined in duplicate by real-time quantitative PCR (qPCR) using the TaqMan ABI 7700 sequencing detection system with heat-activated TaqDNA polymerase and commercial murine primers (Ccl2, Mm00441242_m1; Tnfα, Mm00443258_m1; Il17, Mm00439618_m1; Mmp2, Mm00439498_m1; Nox2, Mm01287743_m1; Nox4, Mm00479246_m1; Fcγr1/Cd64, Mm00438874_m1; Fcγr3/Cd16, Mm00438883_m1; Fcγr4/Cd16.2, Mm00519988_m1; Applied Biosystems), performing an initial step of 2 min at 50 °C and 10 min at 95 °C, and 40 subsequent cycles of 15 s at 95 °C and 1 min at 60 °C. The amplification results of the gene of interest were normalised with the internal control 18S rRNA amplified in parallel. The amount of mRNA was estimated using the 2-ΔCT quantification method.
ELISASyk phosphorylation (p-Syk) levels were determined by cell ELISA. Briefly, cells were seeded in 96-well plates (105 cell/well) and stimulated at different times with IC in the presence or absence of Syk inhibitor. They were then fixed with 4% formaldehyde, blocked (PBS with 2% albumin and 5% FBS) and incubated with anti-p-Syk (Y323, Y317; 44-234G, Invitrogen) or anti-Syk (2712, Cell Signalling) antibody for 16 h at 4 °C, followed by peroxidase-conjugated secondary antibody and the substrate 3,3',5,5'-tetramethylbenzidine. Absorbance was measured on a plate reader (λ = 450 nm and correction to 570 nm). P-Syk values were corrected for total Syk (analysed in parallel wells) and cell content per well (stained with methylene blue). The concentration of CCL2 and TNF-α proteins secreted into the culture medium was quantified using ELISA kits (MJE00, R&D Systems; 88-7324-22, Thermo Fisher Scientific).
Measurement of oxidative stressNADPH oxidase (NOX)-dependent superoxide anion production was determined by chemiluminescence assay, as above.13 Stimulated cells were resuspended in phosphate buffer (50 mM K2HPO4/KH2PO4, .01 mM EDTA,. 32M sucrose, and .1% protease inhibitor cocktail), and transferred to Röhren tubes. The chemiluminescent reaction was initiated by sequential addition of 5 μM of lucigenin and 100 μM of NADPH. Light emission by oxidation of lucigenin was quantified every 10 s for 10 min in a Sirius luminometer (Berthold), expressing the values in chemiluminescence units per milligram of cell protein.
Statistical analysisResults are presented as individual data and mean ± standard deviation (SD) of the total of individual animals or experiments. Statistical analysis was performed with GraphPad v. 8 (GraphPad Software Inc.) using Tukey’s one-way ANOVA test with multiple comparisons. Statistical significance was established for values of P < .05.
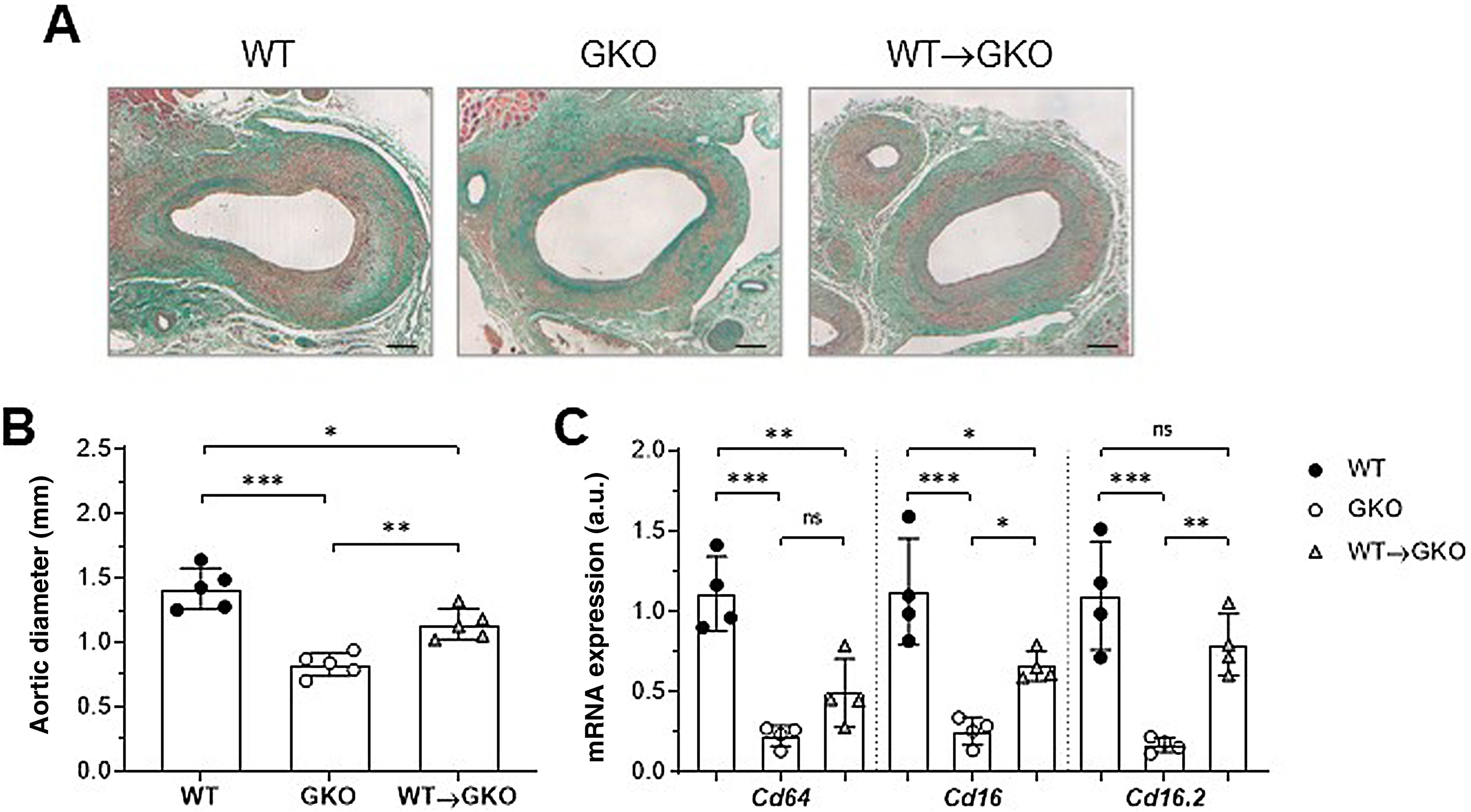
ResultsActivation of FcγR in macrophages aggravates the development of AAA in miceOur previous study demonstrated that the immuno-inflammatory response produced in the aneurysmal lesion promotes the expression of FcγR isoforms in the vascular wall of human AAA and that dysfunction of FcγR protects against the development of experimental AAA.12 To further investigate the specific role of monocyte/macrophage FcγR in AAA formation, we performed a combined model of adoptive transfer and induction of AAA with elastase. For this purpose, bone marrow cells isolated from WT mice were differentiated into macrophages and transferred into GKO mice (deficient in the γ chain associated with FcγRI/CD64, FcγRIII/CD16, and FcγRIV/CD16.2), which were induced by elastase perfusion. After 14 days, histological study was performed on consecutive sections of the abdominal aorta of the mice (Fig. 1A). Compared to the WT group, the GKO mice showed lower incidence of AAA (60% vs. 100%) and smaller aortic diameter (% vs. WT: 42 ± 9; Fig. 1B). In contrast, the GKO mice that received WT macrophages (WT→GKO group) showed 100% AAA incidence and a significant increase in aortic diameter compared to the GKO mice without adoptive transfer (% vs. GKO: 173% ± 28%; Fig. 1B). By qPCR analysis we observed that transfer of WT macrophages partially restored the expression of FcγR activators (Fig. 1C), notably the FcgRIV/CD16.2 isoform, which reached levels close to the WT group.
 of Masson’s trichrome staining (A) and aortic diameter measurement (B) in abdominal aortic sections from wildtype (WT), γ-chain-deficient (GKO), and WT macrophage adoptive transfer (WT→GKO) mice 14 days after elastase aortic perfusion. (C) qPCR analysis of mRNA expression of activating FcγR isoforms. Data normalised by 18S rRNA and expressed in arbitrary units (a.u.). Individual values and mean ± SD of 4–5 mice per group.")
The presence of FcγR activators in macrophages aggravates elastase-induced AAA development in mice. Representative images (scale bars, 100 μm) of Masson’s trichrome staining (A) and aortic diameter measurement (B) in abdominal aortic sections from wildtype (WT), γ-chain-deficient (GKO), and WT macrophage adoptive transfer (WT→GKO) mice 14 days after elastase aortic perfusion. (C) qPCR analysis of mRNA expression of activating FcγR isoforms. Data normalised by 18S rRNA and expressed in arbitrary units (a.u.). Individual values and mean ± SD of 4–5 mice per group.
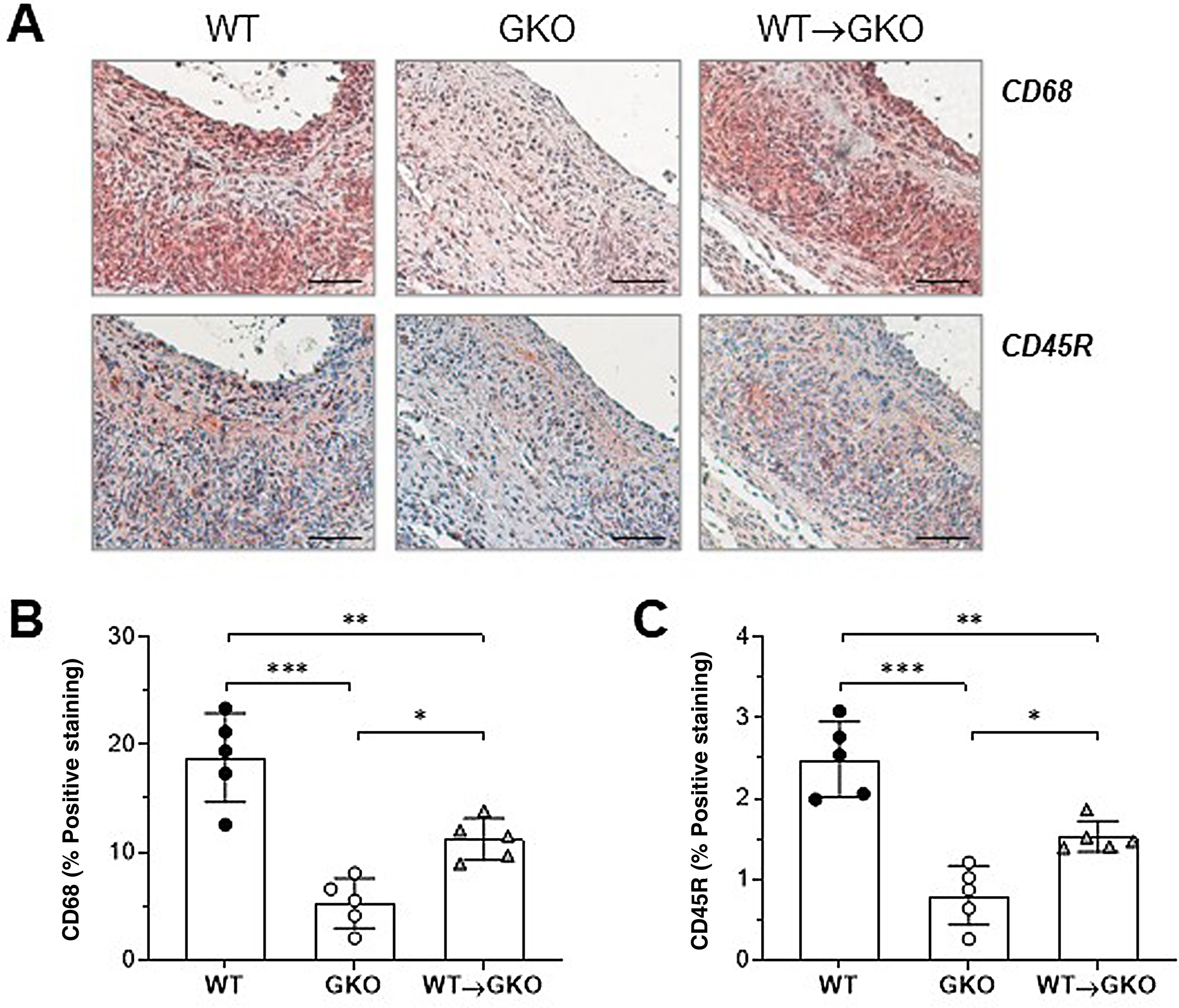
Immunohistochemical analysis of macrophages (CD68 marker) and B lymphocytes (CD45R marker) showed a significant increase in the content of both types of inflammatory cells in the lesions of the WT→GKO group with respect to the GKO group (% vs. GKO: 212 ± 37 and 191 ± 21, respectively), although these values were lower than those of the WT control group (Fig. 2). These results are evidence of an increased inflammatory response in the aneurysmal lesion associated with the presence of macrophages with functional FcγR.
 of immunohistochemical staining (A) and quantification of the area of positive staining of CD68+ macrophages (B) and CD45B+ B lymphocytes (C) in AAA lesions from mice of the WT, GKO, and WT→GKO groups. Individual values and mean ± SD of 5 mice per group.")
Adoptive transfer of macrophages with FcγR activators increases inflammatory infiltrate in AAA lesions. Representative images (scale bars, 50 μm) of immunohistochemical staining (A) and quantification of the area of positive staining of CD68+ macrophages (B) and CD45B+ B lymphocytes (C) in AAA lesions from mice of the WT, GKO, and WT→GKO groups. Individual values and mean ± SD of 5 mice per group.
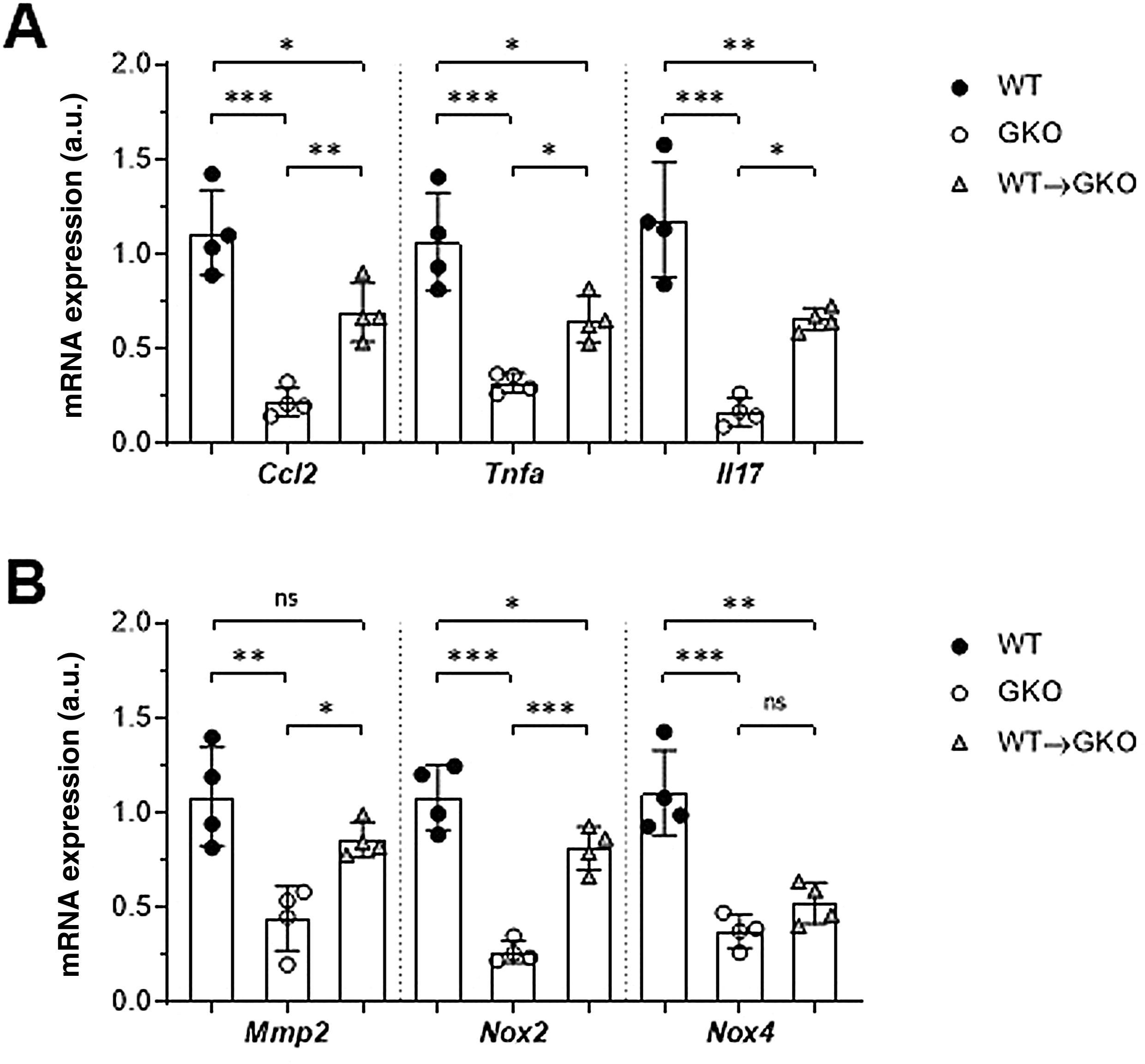
The expression of genes involved in the pathogenesis of AAA was analysed by qPCR. We observed a significant decrease in gene expression of the chemokine CCL2 and the cytokines TNF-α and IL-17 in the abdominal aorta of the GKO mice compared to the WT mice (% vs. WT: 20 ± 7, 30 ± 5, and 14 ± 6, respectively; Fig. 3A). In contrast, the WT macrophage transfer significantly increased these expression levels (% vs. GKO: 314 ± 70, 204 ± 37, and 395 ± 35, respectively; Fig. 3A). In the WT→GKO group, we also observed increased expression levels of MMP2 and the prooxidant enzyme NOX2 (% vs. GKO: 194 ± 21 and 305 ± 43, respectively), whereas the expression of the NOX4 isoform was not significantly changed (Fig. 3B).
 and proteolytic and prooxidant enzymes (B) in murine samples from the WT, GKO and WT→GKO groups. Data normalised by 18S rRNA and expressed in arbitrary units (a.u.). Individual values and mean ± SD of 4 mice per group.")
Impact of transfer of macrophages with FcγR on gene expression in abdominal aorta. qPCR analysis of chemokines and cytokines (A) and proteolytic and prooxidant enzymes (B) in murine samples from the WT, GKO and WT→GKO groups. Data normalised by 18S rRNA and expressed in arbitrary units (a.u.). Individual values and mean ± SD of 4 mice per group.
These results indicate that adoptive transfer of macrophages with functional FcγR in FcγR -deficient mice aggravates the inflammatory and oxidative process and increases susceptibility to the development of AAA.
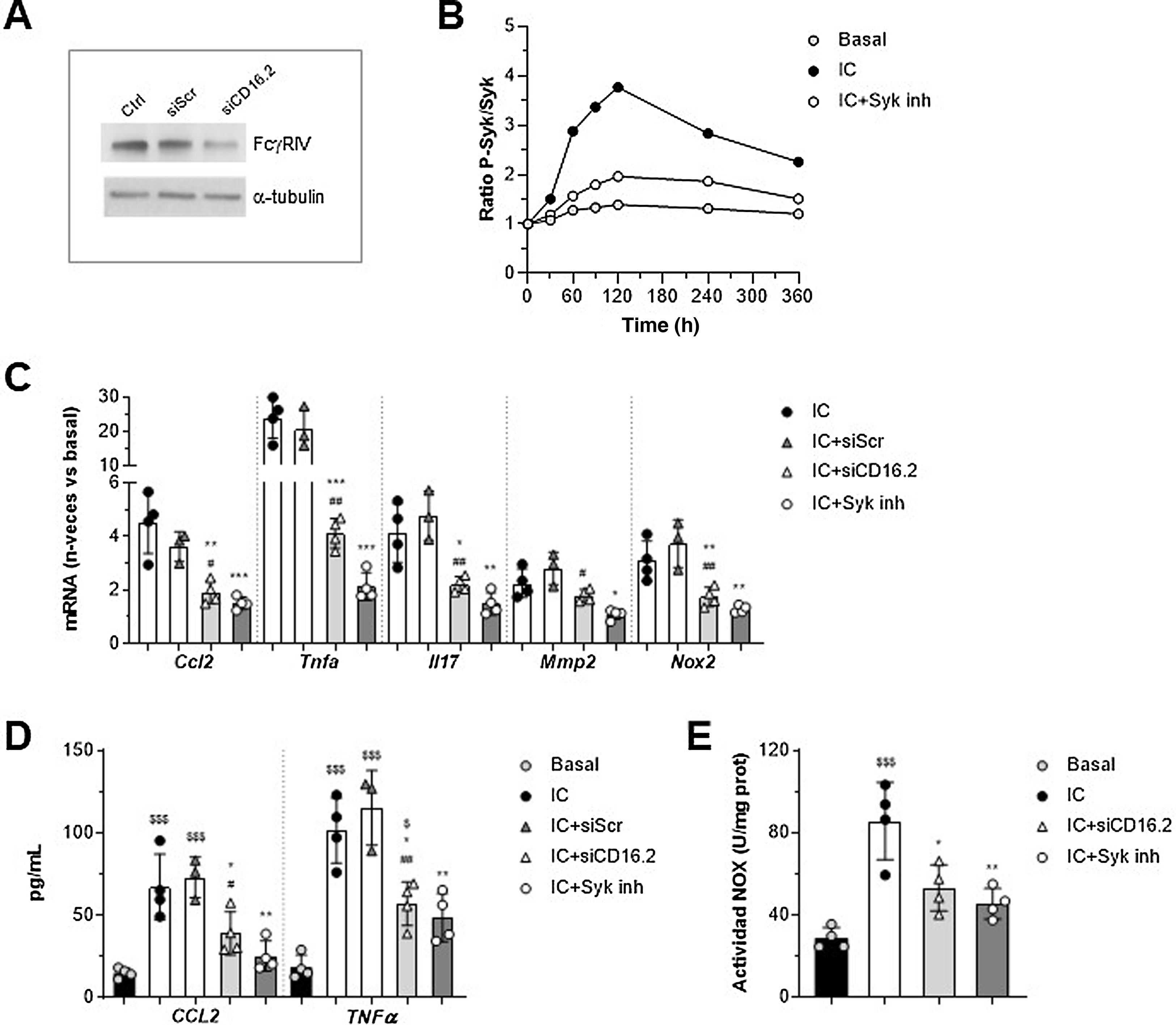
Involvement of FcγRIV/CD16.2 and Syk in in vitro macrophage responseTo further explore FcγR-dependent functions in the context of AAA, we established an in vitro system in murine macrophages stimulated with soluble IgG aggregates as a simile of ICs, as they exhibit similar properties to ICs and exclude the contribution of antigen. On the one hand, since the macrophage transfer mostly restored FcγRIV /CD16.2 levels in aorta, we performed gene silencing experiments of this isoform to determine its contribution to the actions of the IC. Moreover, we evaluated the involvement of Syk tyrosine kinase, a key mediator in the FcγR activator signalling pathway. The suppression of FcγR /CD16.2 in cells treated with ANAip specific for FcgRIV (siCD16.2) compared to the non-specific or scramble condition (siScr) was confirmed by Western blotting (Fig. 4A). By cell-based ELISA we verified that treatment with the selective inhibitor Bay 613606 prevented IC-induced phosphorylation of Syk over time (Fig. 4B).
 and its negative control (siScr), or pre-treated with Bay 613606 inhibitor and subsequently stimulated with IC (IgG aggregates, 150 μg/mL). (A) Representative immunoblot of FcgRIV/CD16.2 and α-tubulin in untransfected (Ctrl) and transfected cells. (B) Cellular ELISA of Syk (phosphorylated/total ratio) in cells treated with Syk inhibitor. Representative experiment performed in quadruplicate. (C) Analysis of mRNA expression by qPCR. Values normalised with 18S rRNA and expressed in increment vs. basal conditions (n = 3–4 experiments). (D) ELISA analysis of CCL2 and TNF-α protein secretion into the cell supernatant (n = 3–4 experiments). Measurement of NOX-dependent ROS production by lucigenin assay, expressed in chemiluminescence units per protein concentration (n = 4 experiments). *P < .05, **P < .01, and ***P < .001 vs. IC; #P < .05 and ##P < .01 vs. siScr; $P < .05 and $$$P < .001 vs. basal.")
Involvement of FcγRIV/CD16.2 and Syk in immunocomplex-mediated responses in vitro. RAW 264.7 macrophages were transfected with FcγRIV/CD16.2 -specific siRNA (siCD16.2) and its negative control (siScr), or pre-treated with Bay 613606 inhibitor and subsequently stimulated with IC (IgG aggregates, 150 μg/mL). (A) Representative immunoblot of FcgRIV/CD16.2 and α-tubulin in untransfected (Ctrl) and transfected cells. (B) Cellular ELISA of Syk (phosphorylated/total ratio) in cells treated with Syk inhibitor. Representative experiment performed in quadruplicate. (C) Analysis of mRNA expression by qPCR. Values normalised with 18S rRNA and expressed in increment vs. basal conditions (n = 3–4 experiments). (D) ELISA analysis of CCL2 and TNF-α protein secretion into the cell supernatant (n = 3–4 experiments). Measurement of NOX-dependent ROS production by lucigenin assay, expressed in chemiluminescence units per protein concentration (n = 4 experiments). *P < .05, **P < .01, and ***P < .001 vs. IC; #P < .05 and ##P < .01 vs. siScr; $P < .05 and $$$P < .001 vs. basal.
qPCR analysis showed a significant reduction of CCL2, TNF-α, IL-17, MMP2, and NOX2 mRNA expression in siCD16.2-transfected and IC-stimulated macrophages compared to untransfected or siScr-transfected cells (Fig. 4C). Similarly, Syk inhibition significantly prevented IC-induced gene expression (Fig. 4C). Both treatments also reduced the secretion of CCL2 and TNF-α proteins into the extracellular milieu of macrophages (Fig. 4D). Finally, by lucigenin assay we observed that incubation of macrophages with IC promotes NOX-dependent superoxide anion generation; this effect was found to be attenuated in cells silenced with siCD16.2 or treated with Syk inhibitor (Fig. 4E).
These in vitro results confirm the direct role of the FcγRIV /CD16.2 isoform and Syk tyrosine kinase in the inflammatory and oxidative response of macrophages to CI stimulation.
DiscussionInflammation, oxidative stress, and extracellular matrix degradation, together with activation of the immune system, are key processes in the pathogenesis of AAA. Several studies show that activation of the autoimmune response to autoantigens deposited or exposed on the vascular wall is involved in the formation and progression of AAA, although the underlying molecular mechanisms are not fully understood.4 The present work demonstrates that IgG FcγR receptors, in particular the activating isoforms present in the macrophage, mediate the immuno-inflammatory response during the development of experimental AAA. Our findings demonstrate that: a) functional deficiency of FcγR activators in GKO mice limits the development of AAA; b) reconstitution with macrophages expressing FcγR aggravates the inflammatory process and formation of AAA, and c) FcγRIV /CD16.2 and Syk molecules are key in the IC-mediated inflammatory and oxidative response in macrophages.
The presence of B-lymphocytes and Ig in AAA lesions is evidence for a role of antibody-mediated humoral immunity in driving AAA formation and progression.14 Several studies have detected elevated levels of IgG, IgA, IgE, and IgM in the serum and/or vascular wall of AAA patients and experimental models.15 An association between plasma and tissue levels of anti-HDL IgG and aortic diameter16 has been found in AAA patients, and circulating levels of antiphospholipid antibodies correlate with markers of inflammation and independently predict the progression of AAA.17 Furthermore, the IgG fraction extracted from AAA lesions shows reactivity to various connective tissue components and is able to activate the complement system, thus participating in the development of AAA.7,18 In mice, B-cell deficiency reduces the presence of Ig in lesions and protects against the development of AAA.19 In this study, we show reduced B-cell infiltration in AAA lesions of GKO-deficient mice and increased infiltration in the mice reconstituted with macrophages expressing FcγR, which is evidence of the involvement of these receptors in the recruitment and/or activation of antibody-producing B cells in the disease.
Previous studies in mice deficient in one or multiple isoforms of FcγR have demonstrated the central role of IgG- FcγR interactions in various autoimmune and inflammatory diseases.8 In cardiovascular diseases, it has been proposed that the generation of autoantibodies directed against self (circulating or vascular wall) epitopes may contribute to vascular inflammation and tissue remodelling through several mechanisms, including opsonisation, and clearance by phagocytes via FcγR, as well as the formation and/or deposition of ICs capable of activating the complement system and the leukocyte and tissue cell response.15 Numerous studies in murine models of atherosclerosis have shown that blockade of activating FcγR isoforms reduces the size of atheromatous plaques,10,11,20 while the absence of the inhibitory receptor aggravates the process.21 However, findings in the context of AAA are more limited. In AAA patients, FcγRIII/CD16 receptor levels in circulating monocytes are associated with levels of inflammatory biomarkers and increased lesion size,22 and in aorta FcγRIIB/CD32B has been found localised in areas of inflammatory infiltrate.23 In our previous study, comparison of human AAA lesions with healthy aortas showed increased expression of all FcγR isoforms. We also detected colocalisation of the activating isoforms FcγRI/CD64 and FcgRIIA/CD16A with IgG deposition in macrophage- and VSMC-rich areas of the vessel wall, showing that both infiltrating and resident vessel cells may participate in the immune response during the formation of AAA.12 In experimental models of AAA, sequencing analysis identified positive regulation of genes associated with the pathway “Fcγ receptor-mediated phagocytosis in macrophages and monocytes”24 and gene deletion of Fc receptors for IgE25 and for IgG12 limits the formation of AAA in models of angiotensin ii or elastase infusion. The findings of the present work indicate that adoptive transfer of macrophages reverses the resistant phenotype of FcγR-deficient mice against the development of AAA, as evidenced by increased AAA lesion size, increased inflammatory cell content, and elevated expression of FcγR isoforms, cytokines, metalloproteinases, and pro-oxidant enzymes. These results highlight the contribution of monocyte/macrophage FcγR to the inflammatory and oxidative response and pathological vascular remodelling during AAA.
Monocyte/macrophage activation is involved in all phases of the formation of AAA, from initial development to aortic dissection or rupture.26 The main factors involved in macrophage accumulation in the AAA wall include chemokines and cytokines produced in response to tissue injury, extracellular matrix degradation products, and ROS production, among others.5 These local microenvironment factors also influence the response and expression profile of macrophages, encouraging their polarisation towards a broad spectrum of phenotypes with both pathogenic and tissue repair functions. During the early stages of AAA lesion formation, there is a gradual increase in macrophages with a proinflammatory M1 phenotype that contributes to oxidative stress, inflammation, and vascular remodelling, while the anti-inflammatory M2 phenotype has been associated with advanced stages, possibly as a compensatory mechanism to prevent AAA expansion and/or rupture.26,27 In human monocytes, various proinflammatory and anti-inflammatory cytokines, as well as the redox state, modulate the balance between activating and inhibitory FcγR and thus the magnitude of their responses.28,29 Moreover, activation of FcγRIIA/CD32A and FcγRIII/CD16 has been reported to promote M1 polarisation,30 while FcγRIIB/CD32B is associated with an anti-inflammatory cytokine-producing M2b phenotype.31 In this work, the adoptive transfer of macrophages with FcγR favours the accumulation of inflammatory cells in the lesions and increases the expression of the chemokine CCL2, the inflammatory cytokines TNF-α and IL-17, and the proteolytic enzyme MMP2, all typical mediators of the M1 inflammatory response and vascular remodelling. We also observed a marked increase in NOX2, the phagocytic isoform mostly involved in ROS generation by macrophages. However, the expression of NOX4, an isoform associated with non-phagocytic cells such as endothelial cells and VSMC, was not altered.32 With this in mind, although our study highlights the importance of monocyte/macrophage FcγR in the formation of AAA, we cannot rule out that other cell types contribute to the pathological process. Indeed, both endothelial cells and VSMC express several isoforms of FcγR, whose stimulation promotes a variety of cellular responses such as cytokine and ROS production, apoptosis, proliferation, and chemotaxis.10,12,33,34
From the perspective of the mechanism, this study implicates the FcγRIV/CD16.2 isoform and Syk kinase in IC-mediated responses in the context of AAA, providing evidence that both molecules could be potential targets for action in the disease. The murine receptor FcγRIV is orthologous to the human FcγRIIIA/CD16A, interacts with intermediate affinity with the most potent IgG subclasses (IgG2a and IgG2b), and is involved in antibody-dependent cellular cytotoxicity and phagocytosis. Mice deficient in FcγRIV/CD16.2 are protected against the development of various immune diseases, while blocking its activity inhibits the release of inflammatory mediators in neutrophils and macrophages.35 Our results demonstrate for the first time that macrophage transfer in vivo restores the expression levels of the FcγRIV/CD16.2 isoform in AAA lesions and that its gene silencing in vitro prevents the expression of proinflammatory and pro-oxidant genes, as well as IC-induced ROS release.
Moreover, aggregation of FcγR activators by IC on the cell surface of leukocytes is known to activate the Src family of tyrosine kinases, which phosphorylates ITAM motifs on the receptor-associated γ chain, promoting Syk phosphorylation and activation, and subsequent activation of multiple signalling pathways.8 Syk is also involved in signalling of B- and T-lymphocyte antigen receptors and C-type lectin receptors, which also contain ITAM motifs and are activated in various immune and inflammatory diseases.36 In AAA samples from patients and experimental models, increased phosphorylation of Syk has been found in the vascular wall.37 In vitro, Syk regulates macrophage differentiation to M1 phenotype, cell proliferation and migration, and secretion of inflammatory mediators,38 and its inhibition suppresses lymphocyte and VSMC responses and limits the progression of AAA.12,37 Confirming these previous publications, our study with the selective Syk inhibitor provides evidence that activation of the FcγR-Syk axis is involved in the inflammatory and oxidative response of macrophages to ICs in the context of AAA.
In conclusion, this work indicates that the interaction between ICs and FcγR activators present in the macrophage participates in the pathogenesis of AAA through the induction of inflammatory mediators, proteolysis, and oxidative stress. These findings support the use of immunomodulatory therapies targeting FcγR or its effector molecules to inhibit humoral immune-mediated vascular damage in AAA.
FundingStudy funded by the Spanish Society of Arteriosclerosis (BIB05/18), the Ministry of Science and Innovation (RTI2018-098788-B-I00 and PID2021-127741OB-I00) and La Caixa Foundation (HR17-00247), and awarded best poster communication at the XXXIV congress of the SEA.
Conflict of interestsThe authors have no conflict of interests to declare.
The authors would like to thank Ana Melgar and Patricia Quesada (IIS-FJD/UAM) for their technical support in the histological studies.