



Omodysplasia, a heterogeneous osteochondrodysplasia, was first observed by Barrow and Fitzsimmons (1984), but better delineated and named by Maroteaux et al. (1989). It was only in 1991, with Borochowitz et al., that two forms were recognized: autosomal dominant and recessive.
Recently, Elçioglu (2004) studied five cases of recessive Omodysplasia and made an excellent review of the literature based on these patients and on 16 other previously reported cases, thus improving knowledge regarding the natural course and variability of the disorder.
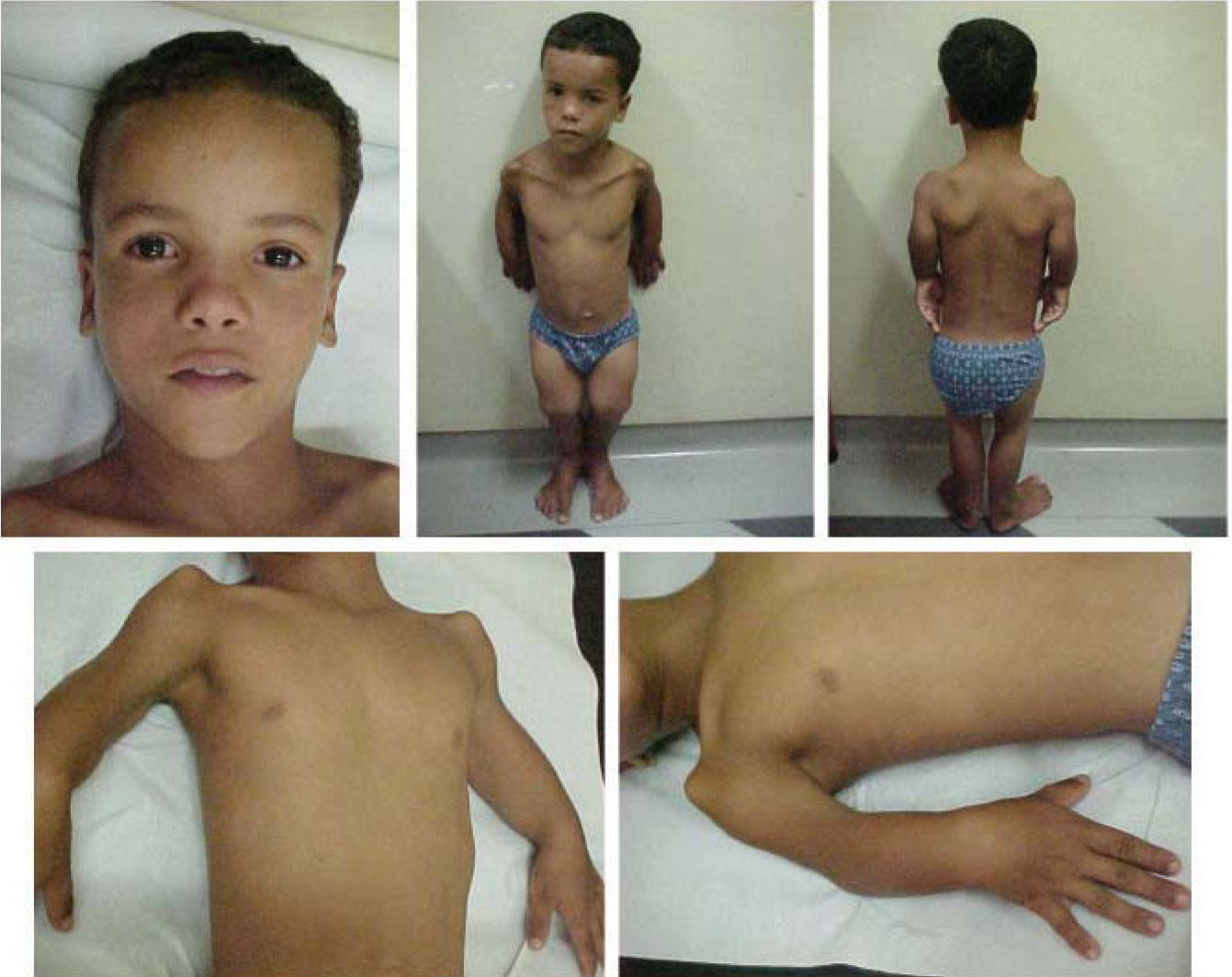
Patients present the following typical clinical and radiological findings: a round and flat facies with a prominent forehead, frontal bossing, short nose with a depressed broad bridge, short columella, anteverted nostrils, long philtrum and small chin. A rhizomelic shortening and radial head dislocation that leads to a characteristic limited extension of the elbows are observed. They also present a short and broad chest, widely spaced nipples and, in males, criptorchidism is frequent. Mental retardation was reported in four patients and delayed motor development occurred in a few cases. Skeletal findings are confined to the limbs with shortened humeri with hypoplastic condyles and axial rotation of their shafts. At birth, radius and ulna are short and thick, but straighten with age. Likewise, in newborns, the humerus is shorter than the radius, but with age it becomes longer. The radial head is anteriorly and laterally dislocated, the ulna is abnormally placed resulting in a radioulnar diastasis. Femora show bulky proximal ends and a distal hypoplasia, remaining relatively short with age, but the distal tapering decreases due to metaphyseal widening and decreasing outward rotation of the femoral shafts. The tibia and fibulae are moderately short and the epiphyseal ossification centers of the femora and tibiae are strikingly flat.
The facial features of omodysplasia overlap with achondroplasia and Robinow syndrome and both these conditions should be considered in the differential diagnosis as well as other ryzomelic dwarfisms. The skeletal survey is decisive for differential diagnosis, because the skeletal defects are characteristic for each syndrome.
In the autosomal dominant form of omodysplasia, which is much rarer than the recessive one, abnormalities are limited to the upper limbs, while in the recessive form there is a generalized involvement of the limbs and severe dwarfism. Both types share the facial features, the typical defect of the distal humerus and the complex deformity of the elbows.
Fewer than 25 autosomal recessive cases have been so far reported. We describe the first Brazilian case, a male patient, born of consanguineous parents, affected by the recessive form of the syndrome.
The genetic cause of omodysplasia has yet to be identified. Omodysplasia shares several phenotypic characteristics with autosomal recessive Robinow syndrome including limb shortening, facial dysmorphisms and genital defects. Recessive Robinow syndrome is caused by mutations in ROR2 (Afzal et al., 2000; van Bokhoven et al, 2000). Considering that both syndromes could affect similar structures, we hypothesized that they could be caused by mutations in the same gene, and tested ROR2 as a candidate for autosomal recessive omodysplasia.
CASE REPORTA 9 year-3 month-old boy with unremarkable family history, complaining of pain in his shoulders, hip and legs was evaluated. He was born at term from healthy consanguineous parents (1st degree), by vaginal delivery, after an uneventful pregnancy. Birth weight was 2,700g and his length at birth is unknown. He presented short stature, frontal bossing, midfacial flattening, depressed nasal bridge, small nose and, shortened arms, limited extension of the elbows and knees, cryptorchidism, and a tiptoe gait. Neuromotor development was within normality.


Mutational screening of ROR2 gene was performed by single strand conformation polymorphism (SSCP) analysis with previously described primers (Van Bohoven et al., 2000).
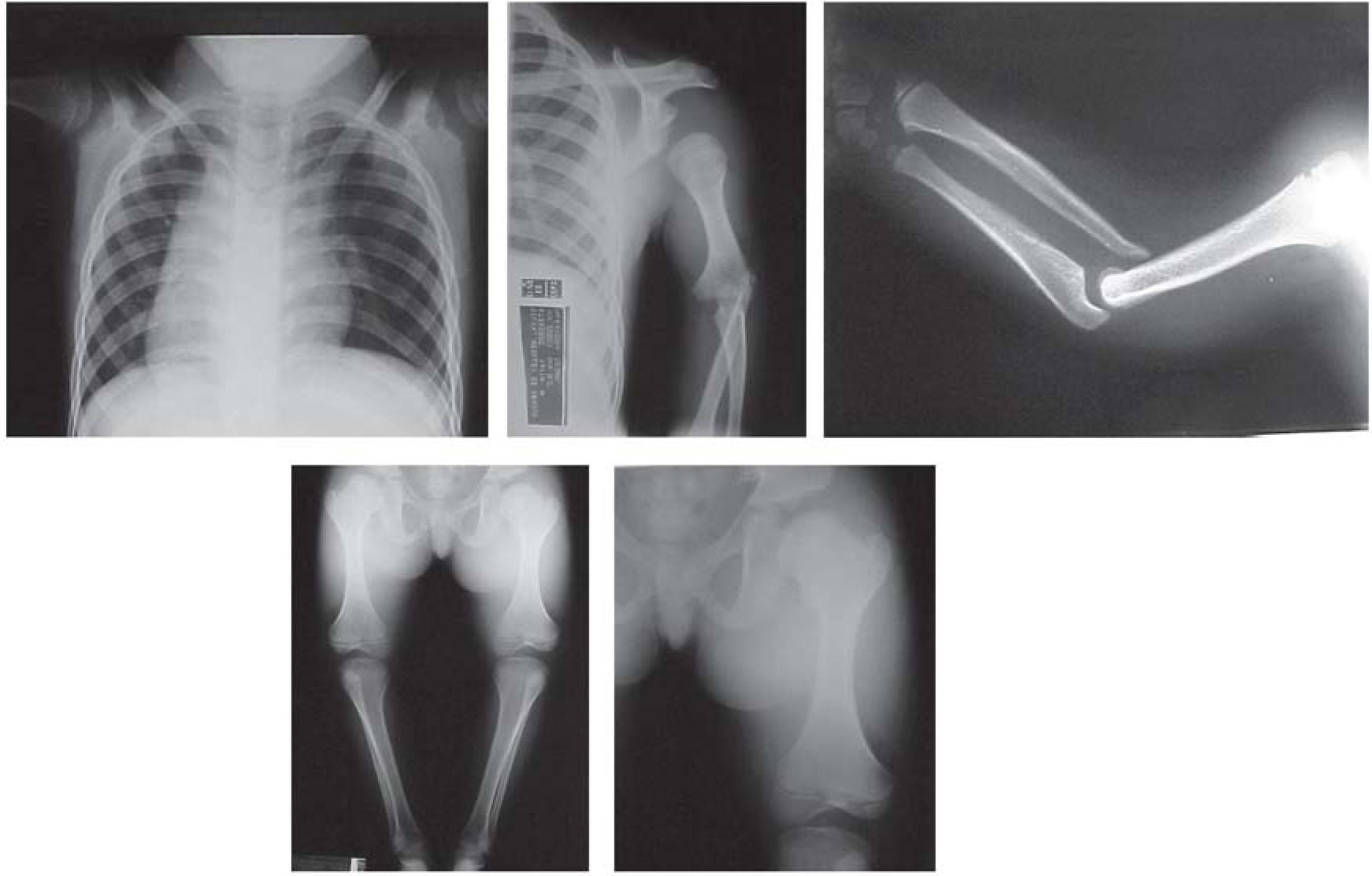
A detailed skeletal survey was performed revealed shortened humeri with abnormal sloping of the distal articular surface, shallow olecranon fossae with partially subluxed radii and radioulnar diastasis; short femora with metaphyseal widening and small laterally displaced patellae. Other bones and growth structures were normal.
Mutation analysis of ROR2 gene by SSCP, showed no altered migration pattern, thus revealing no mutations in these segments.
DISCUSSIONThe patient described herein has the typical features of the autosomal recessive form of Omodysplasia: involvement of the upper and lower limbs and severe short stature. Furthermore, our case is the product of a consanguineous couple.
The features of all autosomal recessive reported cases1–13 were compared with our patient (Table 1). As in other cases of recessive omodysplasia, our patient does not have metacarpal shortening, a feature of the dominant form.
Main features of the reported autosomal recessive cases1-13
| Features | All autosomal recessive reported cases | Presented case |
|---|---|---|
| Sex | 15M/7F | M |
| Characteristic facies | 22/22 | + |
| Limited extension of elbows/knees | 13/21 | + |
| Short humeri | 20/22 | + |
| Short femora | 21/22 | + |
| Dislocated elbows | 22/22 | + |
| Motor delayed/mental retardation | 06/22 | – |
| Short stature | 14/14 | + |
| Cryptorchidism | 12/15 | + |
Our case adds to previous reports that the recessive form of omodysplasia is the most common type with male preponderance, and shows no metacarpal shortening.
In some instances, omodysplasia can be misdiagnosed as recessive Robinow syndrome. Thus, in Robinow syndrome with a predominantly rhizomelic (and not mesomelic, which is typical of Robinow syndrome) limb shortening we can have some difficult. A detailed skeletal survey should then be performed for differential diagnosis. In our patient, limb and genital involvement, resemble those in Robinow syndrome, but the rhizomelic shortening with humerus and radius anomalies are typical of omodysplasia, thus allowing this diagnosis to be established.
Our negative results in the ROR2 gene screening not only reinforce the diagnosis of omodysplasia but also suggest that the two syndromes are not caused by mutations in the same gene, in spite of their phenotypical similarity.
Skeletal survey is an essential tool for the differential diagnosis of omodysplasia, since the skeletal features in this syndrome differ from those in achondroplasia and Robinow syndrome, as well as in other syndromes with ryzomelic disproportion.14,15