





Carney complex is a multiple neoplasia syndrome having endocrine and non-endocrine manifestations. Diagnostic criteria include myxoma, lentigines, and primary pigmented nodular adrenocortical disease, amongst other signs/symptoms.
In most cases it is an autosomal dominant disease, and diagnosis therefore requires study and follow-up of the family members. Inactivating mutations of the PRKAR1A gene were identified as the main cause of the disease, although since 2015 other disease-related genes, including PRKACA and PRKACB activating mutations, have also been related with Carney complex.
This review will address the genetic aspects related to Carney complex.
El complejo de Carney es un síndrome de neoplasia múltiple de tumores endocrinos y no endocrinos, que incluye la presencia de mixoma, lentiginosis cutánea y enfermedad nodular primaria pigmentada, entre otros criterios para el diagnóstico.
En la mayoría de los casos es de transmisión autosómica dominante, por lo que su diagnóstico hace necesario el estudio y seguimiento familiar. Se ha identificado la presencia de mutaciones inactivantes del gen PRKAR1A como causante de la enfermedad. Desde el año 2015 se han agregado otros genes relacionados, como variantes activantes del gen PRKACA y PRKACB.
En este trabajo se ahondará en los aspectos genéticos relacionadas con el complejo de Carney.
The Carney complex (CNC, OMIM160980) was first described in 1985 by J. Aidan Carney as a combination of myxoma, pigmented skin lesions and endocrine axis hyperactivity.1,2 It represents a multiple neoplasm syndrome characterized by the presence of endocrine and non-endocrine tumors. In 70% of the cases CNC exhibits an autosomal dominant hereditary trait with complete penetrance, while in the remaining cases the complex manifests sporadically.3–5
The prevalence of CNC is not clear, due to the low frequency of the disorder. The largest international cohort study published to date describes approximately 750 cases.5,6
The main endocrine manifestation of CNC is primary pigmented nodular adrenal disease (PPNAD).7,8 The latter is a cause of ACTH-independent Cushing's syndrome.9,10
In 2000 CNC was described as being caused by mutations with a loss of function or large deletions of the PRKAR1A gene (OMIM188830) located on chromosome 17q 22_24, which encodes for the type I-alpha regulatory subunit of protein kinase A.11 A second CNC locus was also identified on chromosome 2p16.12 More recently, other mutations have also been described, including defects of the PRKACA and PRKAC9 genes.
A number of genetic and clinical variants have been reported since the initial description of CNC and the identification of the mutations classically related to the disease.
Molecular aspectsThe molecular mechanisms of CNC are related to amplification of the cyclic adenosine monophosphate (cAMP) signal secondary to mutations in components of its intracellular pathway.11
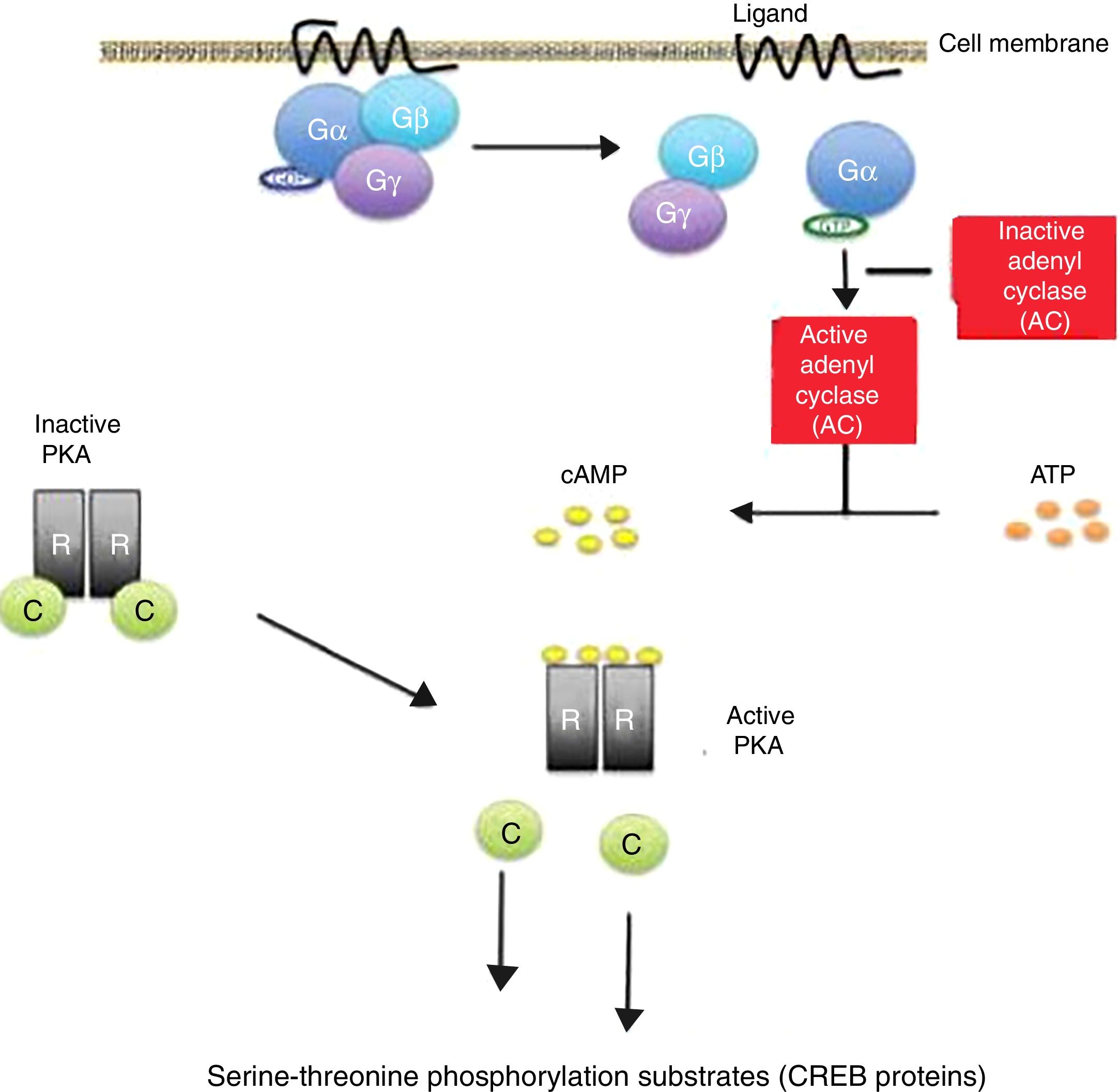
Cyclic AMP induces a phosphorylation cascade which in turn determines modifications in proteins and gene transcription. This molecule is located throughout the human body and is essential for the regulation of many biological functions. Its activation by the alpha (Gα) subunit of cell membrane receptors coupled to protein G triggers the start of its activity in the cells.
The binding of certain ligands or “first messengers” to the membrane receptor associated with protein G leads to an increase in cAMP, which in turn acts as a “second messenger” (Fig. 1). The latter induces a series of changes in cell metabolism that differ according to the type of cell involved.13,14 The cAMP-induced response ends with the mediation of phosphodiesterases, which hydrolyse the molecule.15,16
 activation pathway. Activation of protein G through binding of a ligand stimulates the enzyme adenylate cyclase (AC) to synthesize cAMP. All the genes regulated by cAMP contain a cis action DNA sequence known as the cAMP response element (CRE) that binds the phosphorylated form of a transcription factor called CRE binding protein (CREB). Release of the catalytic subunits (C) of the protein kinase A complex (PKA) is followed by their translocation to the cell nucleus, with phosphorylation of the CREB proteins in the serine-threonine phosphorylation substrates, in response to increased cAMP levels. Inactivating mutations of the PRKAR1A gene in the Carney complex result in the amplification of the cAMP signal. gr1.")
Cyclic adenosine monophosphate (cAMP) activation pathway. Activation of protein G through binding of a ligand stimulates the enzyme adenylate cyclase (AC) to synthesize cAMP. All the genes regulated by cAMP contain a cis action DNA sequence known as the cAMP response element (CRE) that binds the phosphorylated form of a transcription factor called CRE binding protein (CREB).
Release of the catalytic subunits (C) of the protein kinase A complex (PKA) is followed by their translocation to the cell nucleus, with phosphorylation of the CREB proteins in the serine-threonine phosphorylation substrates, in response to increased cAMP levels.
Inactivating mutations of the PRKAR1A gene in the Carney complex result in the amplification of the cAMP signal. gr1.
The protein kinase A (PKA) enzyme complex is the most important intracellular receptor of cAMP in eukaryotic cells. Protein kinase A modulates the activity of several proteins, inducing phosphorylation of the hydroxyl group in the serine and/or threonine groups of different proteins; it therefore constitutes a serine/threonine specific protein kinase.17
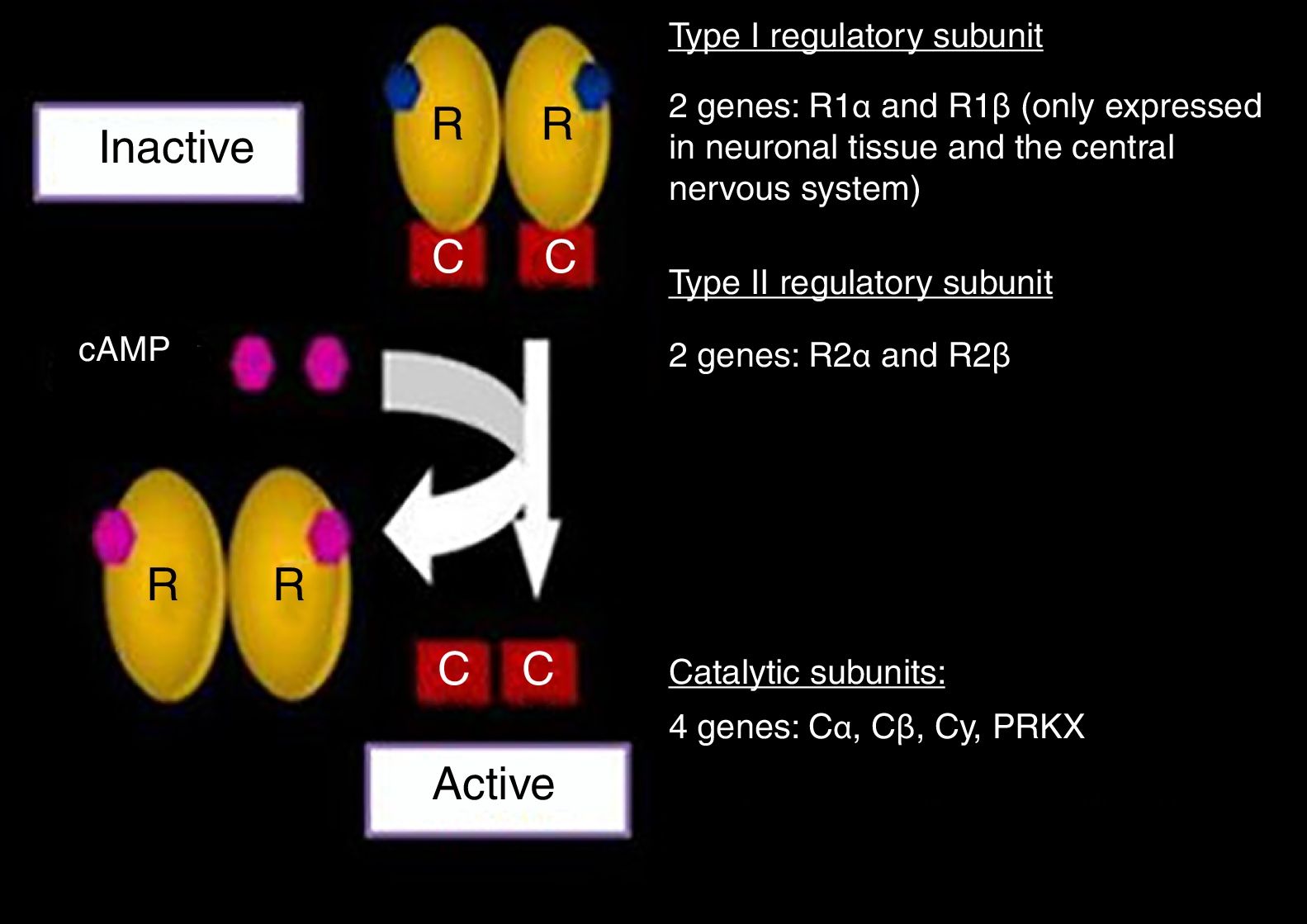
In its inactive state, PKA is a tetrameric complex composed of two regulatory subunits (R): type I-α (RI-α) and type II-β (RII-β), and two catalytic subunits (C): type I-α (CI-α) and type II-β (CII-β). In other words, PKA is a dimer of regulatory subunits bound to two catalytic subunits. Each R subunit has two separate cAMP binding sites. The binding of cAMP to both sites of an R subunit leads to the release of the associated C subunit, revealing its catalytic site and activating its kinase action.18 The best known function attributed to the R subunits is inhibition of the kinase activity of the C subunits.19
The binding of cAMP to an R subunit takes place on a cooperative basis, i.e., the binding of the first cAMP molecule reduces the direct rate constant of the reaction (Kd) for the binding of the second cAMP molecule. In this way, small changes in the cytosolic levels of cAMP can produce major changes in the amount of dissociated catalytic subunits, and thus in the kinase activity of the complex.14,20
The genes encoding for each of the subunits involved in the PKA complex have been studied (Fig. 2). The RI subunit is widely distributed throughout the body, encoded for by the PRKAR1A gene or the gene of the protein kinase A type 1A regulatory subunit. In nerve tissue, this subunit is encoded for by R1 β, which is expressed only in such tissue.11,12,19
 and related genes. Modified and reproduced with permission from Stratakis.22")
Protein kinase A complex (PKA) and related genes. Modified and reproduced with permission from Stratakis.22
The expression of the RII subunit in turn is mediated by the R2 α and R2 β genes. On the other hand, the two catalytic subunits are encoded for by four genes: Ca, Cb, Cy, PRKX. Each one of them, in both its normal and mutated expressions, has been widely investigated.18–25
Activation of genetic transcriptionAll the genes regulated by cAMP contain a cis action DNA sequence known as the cAMP response element (CRE) that binds the phosphorylated form of a transcription factor called CRE binding protein (CREB), which is only found in the cell nucleus. When the PKA C subunits are released in response to increased cAMP levels, they are translocated to the cell nucleus and phosphorylate the CREB proteins in serine-133. The phosphorylated CREB proteins bind to the target genes that contain CRE and also interact with a co-activator called CBP/300, which binds CREB to the basal transcription machinery and allows it to stimulate DNA transcription.14,21
Protein kinase A participates widely in different cell processes, including transcription, metabolism, cell cycle progression and apoptosis. Under normal inactivation conditions, i.e., without binding to cAMP, the four PKA subunits are combined, forming an inactive tetrameric complex. When the regulatory function of the R1 subunit decreases 50%, an increase in the phosphorylation cascade induced by PKA is observed; R1 releases its regulatory action upon the C subunits; and a lack is consequently seen in the control of the kinase activity of these subunits.20–27 In this way, the functional inactivation of the PRKAR1A gene is associated with an excess in the PKA signaling pathway in the affected tissues.
The study of the function of the cAMP-PKA pathway and of the genes that encode it proved crucial for understanding the pathogenesis and genetic findings in patients with CNC. On the other hand, more recent studies have implicated this pathway in other causes of Cushing's syndrome of adrenal origin, from PPNAD and adrenal carcinoma to unilateral cortisol-producing adenomas.28–32
Clinical manifestations of Carney complexThis syndrome is characterized by the presence of endocrine gland and non-endocrine tumors.
The diagnostic criteria were revised in 2001, and in 2015 alterations in other genes apart from PRKAR1A were described (Table 1).
Major diagnostic criteria of the Carney complex.
| 1. Skin lentiginosis with a characteristic distribution (lips, conjunctiva, mucous membranes)a |
| 2. Myxomas (cutaneous and mucous)a or cardiac myxomaa |
| 3. Breast myxomatosisa or MRI findings (with fat suppression) suggestive of the diagnosis |
| 4. Primary pigmented nodular adrenal diseasea or a paradoxical increase in glucocorticoid excretion levels in urine following the administration of dexamethasone |
| 5. Acromegaly associated with growth hormone-producing hypophyseal adenomaa |
| 6. Large-cell calcifying Sertoli cell tumor of the testiclesa or the presence of calcifications in the testicle ultrasound exploration |
| 7. Thyroid carcinomaa or the presence of multiple hypoechoic nodules in the thyroid ultrasound exploration at prepubertal age |
| 8. Psammomatous melanotic schwannomasa |
| 9. Blue nevus, multiple epithelioid blue nevia |
| 10. Multiple ductal adenomas of the breasta |
| 11. Osteochondromyxomaa |
| Complementary criteria |
| 1. Affected first-degree relative |
| 2. Presence of inactivating mutations of the PRKAR1A gene4 |
| 3. Activating variants of the PRKACA gene or PRKACB gene9,30,50 |
| Suggestive findings |
| 1. Multiple pigmented lesions with no typical distribution |
| 2. Multiple blue nevi |
| 3. Cafe au lait or other stains from birth |
| 4. Increase in type 1 insulin growth factor, the oral glucose overload test for abnormal GH or paradoxical GH increase with the TRH test, even in the absence of clinical findings of acromegaly |
| 5. Cardiomyopathy |
| 6. Family history of Cushing's syndrome, acromegaly or sudden death |
| 7. Pilonidal cyst |
| 8. Colon polyps (often associated with acromegaly) |
| 9. Multiple skin moles or other pigmented skin manifestations, multiple lipomas |
| 10. Hyperprolactinemia (often mild and combined with clinical or subclinical acromegaly) |
| 11. Isolated thyroid gland nodule in a patient under 18 years of age, multiple thyroid nodules in a patient over 18 years of age (ultrasound finding) |
| 12. Family history of carcinoma, particularly of the thyroid gland, colon, pancreas and ovary; other multiple benign or malignant tumors |
The diagnosis is based on the presence of two or more major clinical criteria. If the patient has a germinal mutation of PRKAR1A and/or a first-degree relative with CNC, a single clinical manifestation suffices to establish the diagnosis.3,4,9
Lentiginosis of the skin and mucous membranes is the most common presentation of CNC, and the number of lesions typically increases during puberty. The condition exhibits a characteristic distribution around the lips, conjunctiva, and oral and genital mucosa.33
Regarding cardiac myxomas, they manifest at an early age in any heart chamber. They cause obstruction of intracardiac blood flow, embolic phenomena and/or heart failure. Other myxoma locations include the skin, breasts, oropharyngeal region and female genital tract.9
PPNAD causes Cushing's syndrome of adrenal origin and is the most frequent endocrine tumor associated with CNC, being present in 25% of all cases. A paradoxical increase in free cortisol levels in urine in response to the administration of dexamethasone is suggestive of the disorder.29,34 The diagnosis of PPNAD based on imaging techniques remains a challenge, due to the normal appearance of the adrenal glands and the small size of the nodules. The histological study provides confirmation of the diagnosis.9
Over 75% of all males with CNC may present large-cell calcifying Sertoli cell tumors of the testicles. These tumors are generally bilateral and multifocal.9
On the other hand, over 75% of the patients with CNC present multiple thyroid nodules, most of which are related to follicular adenomas.
Psammomatous melanotic schwannoma, a benign neoplastic lesion derived from the Schwann cells, is observed in 10% of the patients with CNC at around 20 years of age.
With regard to hypophyseal lesions, over 75% of all patients with CNC show an asymptomatic increase in growth hormone (GH), type 1 insulin growth factor or prolactin; a detectable tumor is seen in less than 10% of the cases, however. In other words, the biochemical alterations can occur in the absence of a hypophyseal adenoma.35 A single case of corticotropinoma in a patient with CNC has recently been reported.36
Genes related with the Carney complexPRKAR1A geneThe mutations that inactivate the PRKAR1A gene are responsible for the phenotypic manifestations of CNC in over 70% of all cases,5,6 and are classified as type 1 CNC (CNC 1).
These germinal line mutations have also been found in isolated cases of PPNAD (OMIM 610489), with no expression of the remaining manifestations of CNC.31,32
More than 125 different mutations of the PRKAR1A gene have been described to date in over 400 families of different ethnic origins with CNC (http://prkar1a.nichd.nih.gov/hmdb/intro.html).
Most of the mutations result in the appearance of a stop codon. These are “nonsense” mutations that give rise to mRNA encoding for a protein that is shorter than usual (truncated protein). This anomaly in turn is detected by a genetic transcription vigilance mechanism or mRNA nonsense-mediated decay (NMD) system. The system is activated upon the detection of the coding of truncated proteins that would impede normal cell function, and causes the degradation of this RNA. As a result, the anomalous protein is not expressed.6,9,14,24,37 On the other hand, if these changes are located near the end of the gene, the system very possibly will not be activated – thus giving rise to a different phenotype in patients with CNC, as described below.38
Most PRKAR1A inactivating mutations cause a 50% reduction in the expression of protein RI-α, since only the gene in the non-mutated allele is expressed. This phenomenon, known as PRKAR1A haploinsufficiency, results from the incapacity of a single non-mutated gene to maintain the normal phenotype of the individual, and is considered to be the basis of CNC.6,7,11,24,39
Mutations of the PRKAR1A gene with expression at protein level due to a lack of activation of the NMD system are also less frequently observed. These are small insertions and deletions that do not give rise to “frameshift” changes and “splicing” type variants, which do not result in the disease due to protein haploinsufficiency but as a result of the generation of defective proteins that fail to adequately respond to cAMP.38
HotspotsDespite the molecular heterogeneity of CNC, different “hotspots” of the PRKAR1A gene have been identified, where most of the mutations related with the disease are concentrated. For example, the “frameshift” mutation c.491_492delTG (p.Val164Aspfs) in exon 5 was found in over 14 families. Other examples are the deletion c.709-2_709-7delATTTTT found in 11 families, and the single nucleotide variant in exon 2: c.82C>T (p.Gln28Ter), seen in several patients with CNC.5,6,24,39,40
Genotype–phenotype correlationMulticenter studies have demonstrated that certain groups of patients with CNC exhibit clinical characteristics of the syndrome that are correlated to specific mutations. This genetic heterogeneity of the disease and the correlation to certain clinical phenotypes reinforces the importance of the genetic study of patients with CNC. For example, the “hotspot” mutation variant c.491_492delTG (p.Val164Aspfs) is associated with the presence of lentigo, cardiac myxomas and thyroid nodules.5,24,39,40
Patients with PPNAD more often present the mutation c.709-2_709-7delATTTTT and the substitution c.1A>G, which affects the start codon of the protein. It is difficult to correlate the molecular basis underlying this phenotypic expression, though both generate a stop codon and activation of the NMD mechanism.5,6,9
On the other hand, mutations of the PRKAR1A gene that are not related to the NMD mechanism are associated with increased phenotypic expression of all the manifestations of CNC.5,6,9,38
Large deletions of the PRKAR1A gene (from 328bp to 3Mb) have also been reported associated with a more severe and unusual phenotype, possibly due to the haploinsufficiency of additional genes.40
On the other hand, in patients with clinical criteria of CNC without mutation of the PRKAR1A gene, a second CNC locus has been identified, corresponding to a 10Mb region in chromosome 2p16, found by linkage analysis. Tumors in patients with CNC have revealed changes in the number of copies of this region; this group has been referred to as type 2 CNC (OMIM605244). The affected genes at this site have not been identified to date; however, since the CNC1 and CNC2 phenotypes are not significantly different, it has been suggested that the implicated genes could be involved in the same molecular pathway.6,12,24
Phosphodiesterase 11Based on the observation of a degree of correlation between genotype and phenotype in CNC, it has been suggested that certain genes in addition to PRKAR1A could modify the phenotype of the disease. This is the case of the gene encoding for phosphodiesterase 11 (PDE11). This enzyme plays a dual role by catalyzing the hydrolysis of cAMP and cGMP. It is expressed in different organs, though at the adrenal level only the splice A4 variant (PDE11A) is expressed. A decrease in the expression of this gene results in an increase in cAMP, the proteins phosphorylated by the latter, and of the entire cascade induced by it. This happens in the case of mutations that inactivate PDE11A, located on chromosome 2q31_35, and which give rise to a truncated protein.41–47
On the other hand, genetic variants of PDE11A have also been described, with changes in the sequence of the gene that predispose to an association between adrenal and testicular tumors in patients with CNC. Some patients present the coexistence of a mutation of the PRKAR1A gene with these different variants of PDE11A. In such situations a significant association has been observed between the development of PPNAD and testicle tumors (large-cell calcifying Sertoli cell tumors); it is therefore regarded as a phenotype-modifying genetic factor.42–47
Genes encoding for catalytic subunit alpha of the protein kinase A complex (PRKACA)) and for catalytic subunit beta of the protein kinase A complex (PRKACB)
Since 2015, the diagnostic criteria of CNC include the identification of activating variants of the PRKACA and PRKACB9 genes. These are different from the PRKAR1A gene in principle related to the disease, but are also implicated in the cAMP pathway described above.
With regard to PRKACA, which encodes for catalytic subunit C alpha (Cα) of the PKA complex, it was found to be over-expressed in different clinical phenotypes of adrenal disease affecting cortisol synthesis, which are a cause of ACTH-independent Cushing's syndrome, apart from PPNAD classically related to CNC.
The germinal duplication of PRKACA is related to bilateral adrenal hyperplasia, in its different presentations, while the somatic mutations of the same gene give rise to unilateral cortisol-producing adrenal gland adenomas.9,30,31,48,49
In patients with PPNAD as an isolated presentation, the germinal presence of gains in the number of copies (duplication) of the genomic region of chromosome 19, which includes the PRKACA gene, has been determined. On the other hand, its somatic mutation c.617A>C (Leu206Arg) was identified in patients with cortisol-producing adrenal gland adenomas.30,31,48,49In vitro studies determined that this mutation results in the constitutive activation of Cα, with a deterioration in the inhibition of the regulation of both catalytic subunits, exerted under normal conditions, by the PKA regulatory subunits. In addition, an increase in the protein levels of the catalytic subunits of the PKA complex was confirmed in patients with gains in the number of copies of chromosome 19.30
On the other hand, with regard to the PRKACB gene, its over-expression was identified in a patient with CNC who had no mutation of the PRKAR1A gene or alteration of PRKACAThis case corresponded to a 19-year-old woman with manifestations of CNC that included acromegaly, pigmented skin lesions and myxomas, with no Cushing's syndrome. The genomic study identified a 1.6Mb triplication of chromosome 1p31.1 that included the PRKACB gene, which encodes for catalytic subunit beta (Cβ) – the second most important catalytic subunit of PKA. The defect was confirmed by in situ hybridization, which revealed additional genetic material in the supernumerary chromosome. The Cβ levels, but not those of Cα, were found to be increased in lymphocytes and fibroblasts of the patient, and in the material of a breast myxoma. The lymphocytes also showed increased PKA activity with levels similar to those found in other patients with CNC related to inactivating mutations of the PRKAR1A50 gene.
ConclusionCarney complex is a rare autosomal dominant hereditary disorder. Genetic study of the index cases and of the first-degree relatives is therefore important.
As with all infrequent diseases, it is important to clinically monitor the patients and to investigate the molecular basis of the disorder through multicenter studies, in order to obtain more in-depth knowledge of those aspects of this syndrome which remain unclear.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Bosco Schamun MB, Correa R, Graffigna P, de Miguel V, Fainstein Day P. Revisión del complejo de Carney: Aspectos genéticos. Endocrinol Diabetes Nutr. 2018;65:52–59.
