




To assess the frequency and the clinical, biochemical, and molecular aspects of familial hypercholesterolemia (FH) in subjects attending an endocrinology unit.
MethodsAn observational, descriptive study evaluating 3140 subjects attending the endocrinology unit of Centro Médico Orinoco in Ciudad Bolívar, Venezuela, from 7 January 2013 to 9 December 2016. The index cases were selected using the Dutch Lipid Clinic Network criteria. Plasma lipid levels were measured, and a molecular analysis was performed by DNA sequencing of the LDLR and APOB genes.
ResultsTen (0.32%) of the 3140 study patients had clinical and biochemical characteristics consistent with FH. All but one were female. Three had first-degree relatives with prior premature coronary artery; and none had a personal history of this condition. Three patients were obese; three had high blood pressure; and no one suffered from diabetes. Three patients had a history of tendon xanthomas, and one of corneal arcus. LDL-C levels ranged from 191 to 486mg/dl. Two patients were on statin therapy. The genetic causes of FH were identified in four patients, and were LDLR gene mutations in three of them and an APOB gene mutation in exon 26 in the other.
ConclusionApproximately, one out of every 300 people attending this endocrinology unit in those four years had FH, and LDLR gene mutations were the most prevalent cause.
Describir la frecuencia, los aspectos clínicos, bioquímicos y moleculares de la hipercolesterolemia familiar (HF) en sujetos que acuden a una unidad de endocrinología.
MétodosEstudio observacional, descriptivo en el que se evaluaron 3.140 sujetos que fueron atendidos en la Unidad de Endocrinología del Centro Médico Orinoco en Ciudad Bolívar, Venezuela, desde el 7 de enero del 2013 al 9 de diciembre del 2016. Los casos índice fueron seleccionados de acuerdo con los criterios de la Red de Clínicas de Lípidos de Holanda. Se midieron lípidos plasmáticos. El análisis molecular se realizó por medio de secuenciación de ADN de los genes LDLR y APOB.
ResultadosDe los 3.140 sujetos evaluados, 10 (0,32%) tuvieron características clínicas y bioquímicas compatibles con HF. Todos, excepto uno, eran de sexo femenino. Tres pacientes tuvieron antecedente familiar en primer grado de enfermedad coronaria prematura y ninguno antecedente personal de esta patología. Tres pacientes tuvieron obesidad, 3 hipertensión arterial y ninguno tuvo diabetes. Tres pacientes presentaban xantomas tendinosos y solo uno arco corneal. Los valores de c-LDL oscilaron entre 191 y 486 mg/dl. Solo 2 recibían tratamiento con estatinas. En 4 pacientes se identificó la causa genética de la HF: 3 de ellos por mutaciones en el gen LDLR y uno por mutación en el exón 26 del gen APOB.
ConclusiónAproximadamente una de cada 300 personas que acuden a consulta en esta unidad de endocrinología presentan HF. Las mutaciones en el gen LDLR son las principales causantes de HF en este grupo de pacientes.
Familial hypercholesterolemia (FH) (OMIM 143890) is the monogenic disorder most frequently associated to premature coronary disease, due to high concentrations of low density lipoprotein cholesterol (LDLc).1 The disease exhibits an autosomal dominant hereditary trait and is mainly caused by mutations of the LDLc receptor gene (LDLR) and, less frequently, by mutations of the genes encoding for apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9).2,3
Heterozygous FH affects approximately one out of every 300–500 individuals in the general population.4 In turn, homozygous FH affects one out of every 1,000,000 individuals, though its prevalence is greater in certain regions or countries, presumably as a consequence of high endogamy rates.5
People with heterozygous FH have a three- to four-fold higher risk of suffering coronary disease, and on average tend to develop it a decade earlier than in the general population.3,6 Consequently, the early identification of individuals with FH is crucial, since early treatment can lower the risk of premature atherosclerosis.7 However, most patients are neither diagnosed nor treated.8
The prevalence of FH in Latin America is not known, and in the concrete case of Venezuela no previous studies have evaluated the frequency of the disease or the clinical characteristics of the affected subjects.9 To the best of our knowledge, only one study, conducted in Maracaibo, Estado Zulia, has investigated the presence of mutations in exon 4 of the LDLR gene in 65 hypercholesterolemic individuals without a clinical diagnosis of FH, with the identification of mutations in 5 patients.10 There is a lack of knowledge of the disease among the medical community in general in this country. This fact, and the lack of public policies allowing an adequate registry of patients with FH, together with the absence of reference centers specialized in lipid disorders, limit the diagnosis and opportune management of the disease. The present study was therefore carried out to describe the frequency, clinical aspects and biochemical features of FH in individuals seen in an endocrinology unit in Ciudad Bolívar, Venezuela, and to characterize the mutations capable of producing FH in these patients.
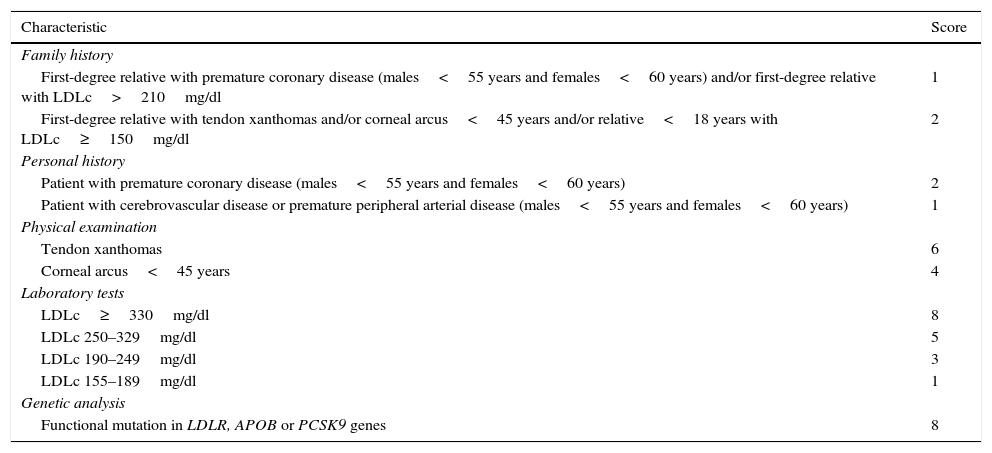
MethodsStudy design and subjectsA descriptive observational study was carried out involving 3140 subjects seen in the Unit of Endocrinology, Diabetes, Metabolism and Nutrition of Centro Médico Orinoco in Ciudad Bolívar (Venezuela), between 7 January 2013 and 9 December 2016. The index cases with clinically and biochemically suspected FH were selected based on the criteria of the Dutch Lipid Clinic Network (DLCN) (Table 1).11 The diagnosis is considered to be firm when the score is >8 points and probable when the score is 6–8 points.11,12 The clinical diagnosis of FH included both groups, since it is possible to detect mutations that cause FH in a significant number of subjects with a probable diagnosis. The molecular analysis included patients with a score of ≥6 points, as recommended by the European Atherosclerosis Society8 and the consensus on the diagnosis and management of FH in Spain.12
Criteria of the Dutch Lipid Clinic Network (DLCN) for the diagnosis of familial hypercholesterolemia.
| Characteristic | Score |
|---|---|
| Family history | |
| First-degree relative with premature coronary disease (males<55 years and females<60 years) and/or first-degree relative with LDLc>210mg/dl | 1 |
| First-degree relative with tendon xanthomas and/or corneal arcus<45 years and/or relative<18 years with LDLc≥150mg/dl | 2 |
| Personal history | |
| Patient with premature coronary disease (males<55 years and females<60 years) | 2 |
| Patient with cerebrovascular disease or premature peripheral arterial disease (males<55 years and females<60 years) | 1 |
| Physical examination | |
| Tendon xanthomas | 6 |
| Corneal arcus<45 years | 4 |
| Laboratory tests | |
| LDLc≥330mg/dl | 8 |
| LDLc 250–329mg/dl | 5 |
| LDLc 190–249mg/dl | 3 |
| LDLc 155–189mg/dl | 1 |
| Genetic analysis | |
| Functional mutation in LDLR, APOB or PCSK9 genes | 8 |
Pregnant women were excluded from the study, in the same way as individuals with a history of alcohol abuse, patients with human immunodeficiency virus (HIV) infection subjected to antiretroviral therapy, subjects with cholestasis or liver failure, endocrine disorders such as untreated hypothyroidism and Cushing syndrome, patients diagnosed with other primary hyperlipidemias, individuals with a triglyceride concentration ≥400mg/dl, and subjects receiving androgens, cyclosporine, amiodarone, retinoic acid and corticosteroids. The study was approved by the local Ethics Committee, following the guidelines of the Declaration of Helsinki, and all subjects gave written consent to participation in the study.
Anthropometric and clinical variablesWeight and height were measured under fasting conditions and with subjects in underwear. Body mass index (BMI) was calculated as weight divided by squared height. Obesity was defined as BMI ≥30kg/m2.13 In subjects with suspected FH, we evaluated the tendons—fundamentally the Achilles tendon and the extensor tendons of the hands—in order to identify possible xanthomas, and the periphery of the iris was examined to identify a possible corneal arcus.
Biochemical variablesA blood sample was obtained from a vein in the forearm after a fasting period on no less than 8h. Total cholesterol, triglycerides and high density lipoprotein cholesterol (HDLc) were quantified via enzyme methods using a Hitachi 911® autoanalyzer and reagents supplied by Cienvar. Low density lipoprotein cholesterol in turn was determined from the Friedewald equation, where LDLc=total cholesterol−[HDLc+(triglycerides/5)].
Genetic analysisThe genetic analysis was carried out using DNA samples extracted from peripheral blood, using the Wizard® genomic DNA purification kit (Promega, USA). The genetic diagnosis of FH was established in two phases: phase 1 included the study of the most frequent APOB gene mutations (fragments of exons 26 and 29) and the molecular study of the promoter, splicing and the encoding regions of the LDLR gene, while phase 2 involved the study of major rearrangements using the multiplex ligation-dependent probe amplification (MLPA) technique.
Data presentationThe patient characteristics are reported in the form of tables and figures, while the clinical and laboratory test data are presented individually.
ResultsThe study sample consisted of 3140 individuals seen in the Unit of Endocrinology, Diabetes, Metabolism and Nutrition. Of these subjects, 10 (0.32%) presented clinical and biochemical characteristics consistent with FH. Five of these 10 patients had a clinical diagnosis of probable FH, while the remaining 5 had a firm diagnosis of FH (score >8).
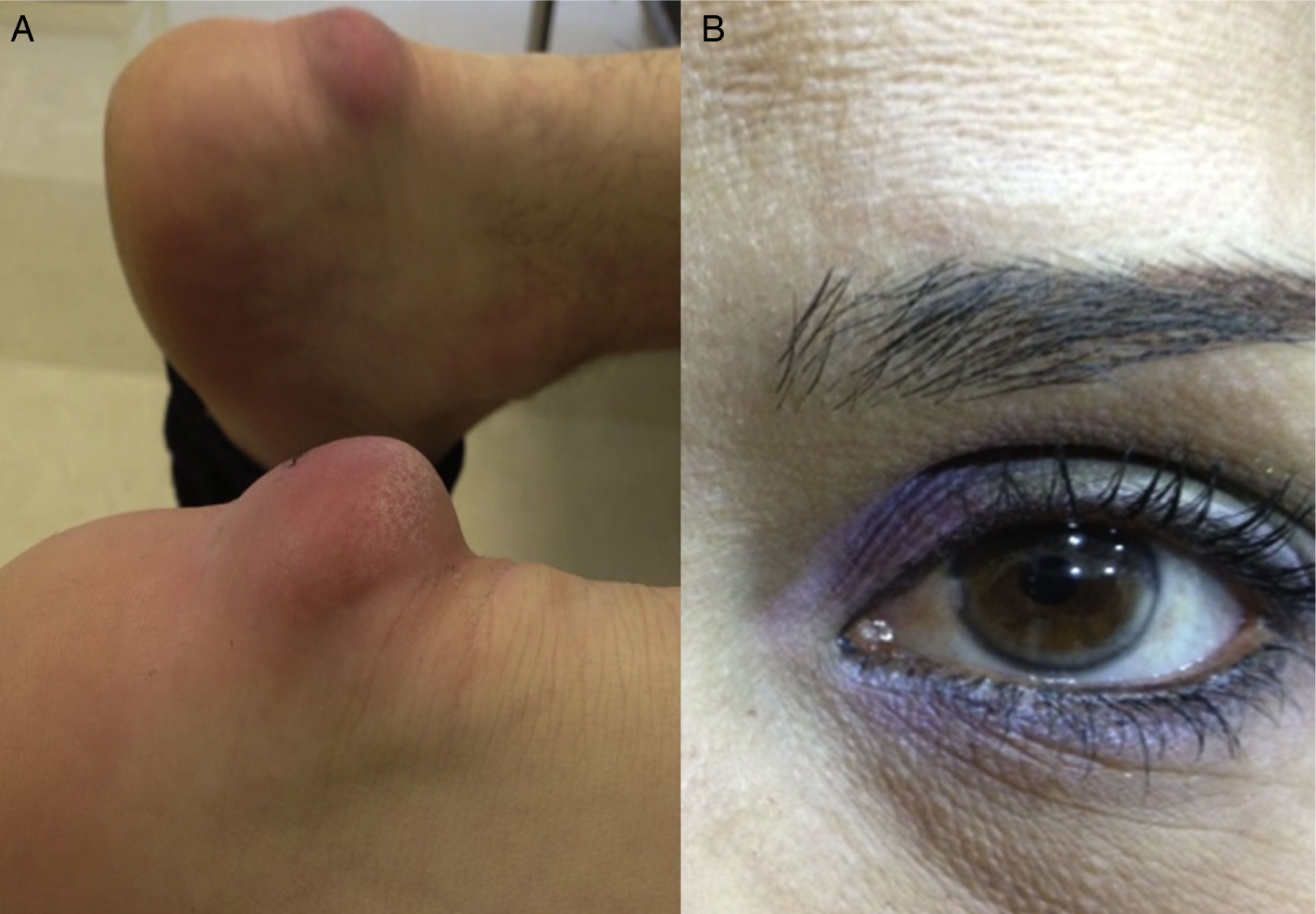
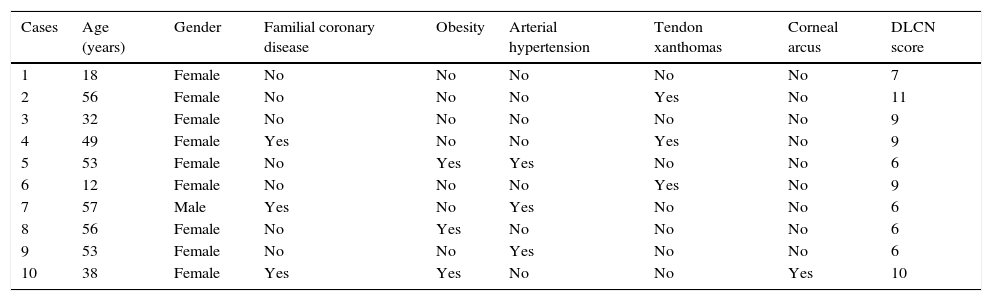
Table 2 shows the clinical data of the 10 patients, with ages between 12 and 57 years. All but one were females. Three patients had some first-degree relative with premature coronary disease, while none of them had a personal history of such disease, peripheral arterial disorders or cerebrovascular disease. With regard to comorbidities, three patients were obese and three had primary arterial hypertension. None suffered diabetes. In relation to the clinical manifestations associated to FH, three patients presented tendon xanthomas, and only one was diagnosed with corneal arcus. Fig. 1 shows the Achilles tendon xanthomas in patient 2 (aged 56 years) and the corneal arcus of patient 10 (aged 38 years).
Clinical data of the patients with familial hypercholesterolemia.
| Cases | Age (years) | Gender | Familial coronary disease | Obesity | Arterial hypertension | Tendon xanthomas | Corneal arcus | DLCN score |
|---|---|---|---|---|---|---|---|---|
| 1 | 18 | Female | No | No | No | No | No | 7 |
| 2 | 56 | Female | No | No | No | Yes | No | 11 |
| 3 | 32 | Female | No | No | No | No | No | 9 |
| 4 | 49 | Female | Yes | No | No | Yes | No | 9 |
| 5 | 53 | Female | No | Yes | Yes | No | No | 6 |
| 6 | 12 | Female | No | No | No | Yes | No | 9 |
| 7 | 57 | Male | Yes | No | Yes | No | No | 6 |
| 8 | 56 | Female | No | Yes | No | No | No | 6 |
| 9 | 53 | Female | No | No | Yes | No | No | 6 |
| 10 | 38 | Female | Yes | Yes | No | No | Yes | 10 |
DLCN: Dutch Lipid Clinic Network.
 Achilles tendon xanthomas in a 56-year-old woman. (B) Corneal arcus in a 38-year-old woman.")
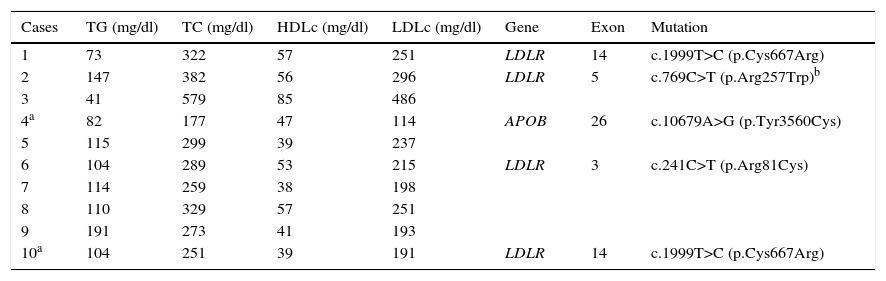
Table 3 shows the data referred to lipid parameters and mutations of the LDLR and APOB genes in the 10 patients with FH. Although two patients were receiving statin therapy, only one of them (patient 4) had acceptable LDLc levels (114mg/dl). The rest of the patients presented clearly elevated LDLc concentrations between of 191 and 486mg/dl. Most of the subjects had normal triglyceride and HDLc concentrations. A total of four different mutations were identified: three in the LDLR gene and one in the APOB gene. The variant p.Cys667Arg was the most common presentation, being observed in two unrelated patients, while the remaining mutations—p.Arg81Cys, p.Arg257Trp and p.Tyr3560Cys (the latter corresponding to the APOB gene)—were identified in a single individual. On the basis of these data, and considering that the p.Arg257Trp mutation has been classified as non-pathogenic in a functional study, the molecular diagnosis was confirmed in 40% of the patients with FH.
Lipid and LDLR and APOB gene mutation profiles in patients with familial hypercholesterolemia.
| Cases | TG (mg/dl) | TC (mg/dl) | HDLc (mg/dl) | LDLc (mg/dl) | Gene | Exon | Mutation |
|---|---|---|---|---|---|---|---|
| 1 | 73 | 322 | 57 | 251 | LDLR | 14 | c.1999T>C (p.Cys667Arg) |
| 2 | 147 | 382 | 56 | 296 | LDLR | 5 | c.769C>T (p.Arg257Trp)b |
| 3 | 41 | 579 | 85 | 486 | |||
| 4a | 82 | 177 | 47 | 114 | APOB | 26 | c.10679A>G (p.Tyr3560Cys) |
| 5 | 115 | 299 | 39 | 237 | |||
| 6 | 104 | 289 | 53 | 215 | LDLR | 3 | c.241C>T (p.Arg81Cys) |
| 7 | 114 | 259 | 38 | 198 | |||
| 8 | 110 | 329 | 57 | 251 | |||
| 9 | 191 | 273 | 41 | 193 | |||
| 10a | 104 | 251 | 39 | 191 | LDLR | 14 | c.1999T>C (p.Cys667Arg) |
APOB: apolipoprotein B gene; HDLc: high density lipoprotein cholesterol; LDLc: low density lipoprotein cholesterol; TC: total cholesterol; LDLR: low density lipoprotein receptor gene; TG: triglycerides.
To the best of our knowledge, this is the first study in a Venezuelan endocrinology unit to not only evaluate the frequency of FH but also the clinical, biochemical and molecular characteristics of the affected individuals. The results of the study suggest that approximately one out of every 314 individuals seen in an endocrinology unit present FH. Heterozygous FH is commonly assumed to affect one out of every 500 individuals in the general population.14 However, recent reports suggest an even higher prevalence of approximately one case out of every 200–300 persons,3,4,8 which coincides with the observations of our study.
Tendon xanthomas are pathognomonic of FH, and were seen in 30% of our patients. Individuals with xanthomas have been shown to suffer a three-fold higher risk of coronary disease than subject without xanthomas.15 Despite their clinical usefulness, xanthomas are observed in less than 30% of all cases. Their absence therefore does not rule out a clinical diagnosis of FH.16
Worldwide, most patients with FH are not diagnosed and therefore receive no treatment, or only insufficient treatment.8,17 The detection of FH complies with the criteria of the World Health Organization (WHO) for systematic screening of a disease, and is cost-effective in identifying new cases of FH through familial cascade screening from identification of the index cases.12 In our study we found that none of the patients with FH had been previously diagnosed, and only two had received lipid-lowering treatment with statins—though without reaching the therapeutic control targets (LDLc<100mg/dl). Of note is the observation that none of the treated subjects received combination therapy or maximum dose statin treatment. This coincides with the findings of a Spanish observational study involving the participation of primary care physicians, in which less than 5% of the cases with a genetic diagnosis reached the LDLc targets, and less than 15% of these patients were receiving maximum dose combination therapy.16
The criteria of the Dutch Lipid Clinic Network (DLCN) were developed with the purpose of facilitating the screening of patients for genetic analysis, as part of the national FH screening program in The Netherlands.11 Based on these criteria, it is seen that a clinical, biochemical and molecular diagnosis could be established in 40% of the selected patients (probable+firm diagnosis), since the mutation causing FH was identified—the detection rate increasing with the score obtained. These results are similar to those reported in Denmark, where mutations were detected in 48.1% of the subjects on including those with a probable and a firm diagnosis. In comparison, the detection rate increased to 62.9% on only considering those patients with a firm diagnosis.18
In concordance with other Latin American countries and the rest of the world,2,9 most of the mutations found in patients with FH affect the LDLR gene, located on the short arm of chromosome 19 and composed of 18 exons.19 Over 1800 different variants have been described to date, not all of which are pathogenic.20 Based on the location of the mutations, 5 classes affecting different sites within the LDLc receptor pathway have been identified: class 1 (lack of synthesis of the LDLc receptor), class 2 (receptor transport defect), class 3 (lack of LDLc binding to its receptor), class 4 (altered LDLc/LDLc receptor complex internalization) and class 5 (receptor recycling dysfunction).19
A study carried out in Maracaibo (Venezuela) evaluated a total of 65 patients with elevated LDLc and analyzed exon 4 of the LDLR gene and mutations R3500Q, R3500W and R3500C of the APOB gene. None of the investigated individuals had tendon xanthomas or corneal arcus, and only one presented xanthelasma. An LDLR mutation was identified in 5 patients: four were novel while one had been previously described in a German population, likewise associated to hypercholesterolemia.10 It should be noted that none of these mutations were observed among patients in Ciudad Bolívar.
In index cases 1 and 10 (unrelated individuals) we identified the same mutation: thymine substituted by cytosine in nucleotide 1999, resulting in the substitution of cysteine by arginine in codon 667 of exon 14 of LDLR. This mutation has been previously described in relatives of a family in Syria, where this substitution was shown to be able to affect transport of the LDLc receptor between the endoplasmic reticulum and the Golgi complex.21
The molecular analysis of patient 2 identified the variant c.769C>T, located in exon 5 of the LDLR gene. This variant resulted in the substitution of arginine by tryptophan in position 257 of the LDLc receptor. This alteration is subject to controversy, since it has been described at least three times in families with FH.22–24 However, recent functional studies have indicated that it is a non-pathogenic variant.25 In turn, patient 6 was found to have mutation c.241C>T, located in exon 3 of the LDLR gene, which generates the substitution of arginine by cysteine in position 81 of the LDLc receptor. This antisense mutation has been previously described and affects the receptor binding domain.26 This was the only patient under 18 years of age included in the study, and FH was suspected due to the presence of LDLc>190mg/dl, as suggested by some diagnosis and treatment guides.11,12
A mutation of the APOB gene is found in 5% of all patients diagnosed with FH.27 This disorder is also known as familial defective apolipoprotein B and produces a phenotype identical to that of FH. Patient 4 was found to have mutation c.10679A>G, located in exon 26 of the APOB gene, which generates the substitution of tyrosine by cysteine in position 3560 of apolipoprotein B. This mutation has also been previously described28 and affects LDLc particle binding to its receptor—giving rise to an increase in plasma LDLc concentration.
In patients with clinically diagnosed FH in which no mutation is detected, the increase in plasma LDLc concentration may be due to polygenic causes, i.e., an accumulation of genetic variants that increase LDLc and can yield a phenotype similar to that of FH.29 Another possible explanation is the impossibility of the current techniques to detect all the mutations, e.g., intronic mutations in LDLR, mutations in PCSK9 or mutations in other undiscovered genes that cause FH.30 It should be mentioned that the PCSK9 gene was not analyzed in our study, though independently of the detection of mutations, the diagnosis of FH should not be ruled out, since no monogenic defects have been identified in a significant number of cases in which the main FH-causing genes were investigated.30 Furthermore, all patients should be adequately treated, since most morbidity-mortality studies have been conducted in patients with a clinical diagnosis, not a genetic diagnosis.31,32 In any case, it has been shown that for any LDLc concentration, established genetic mutation carriers are at an increased risk of suffering coronary disease.33
Patients with FH have a high risk of developing cardiovascular diseases. However, this risk can be modified by the presence or absence of other concomitant factors such as diabetes mellitus, arterial hypertension and obesity.8,12 It should be mentioned that none of the analyzed patients with FH had a personal history of coronary or cerebrovascular disease. This could be due to the presence of favorable factors such as the female gender (90% of the cases), an age of under 40 years (40%) and the absence of smoking in all subjects.
Study limitationsAlthough our study offers data of interest, it admittedly has some limitations: (1) The frequency of FH was investigated in individuals seen in an endocrinology unit. As a result, the findings may not be representative of the general population in Ciudad Bolívar or in Venezuela as a whole. Nevertheless, many prevalence studies in the literature are based on hospital registries, hospitalized patient series and even calculations using the Hardy–Weinberg equation and the estimated frequency of homozygous FH.2,30 (2) Some patients visiting the clinic receive lipid-lowering treatment, and the diagnosis of FH proves more complex in the absence of a family history or clinical evidence of hypercholesterolemia. As a result, with this screening method some individuals with FH might not have been selected.
ConclusionsIn conclusion, FH is a worldwide public health problem in view of its frequency and the seriousness of its consequences. In our Unit of Endocrinology, Diabetes, Metabolism and Nutrition of Ciudad Bolívar (Venezuela) we identified one case of FH out of every 314 patients. This suggests important under-recording of the disease and underscores the shortcomings of our health system in detecting patients with FH. Mutations affecting the LDLR gene are the main cause of FH in this group of patients.
Conflict of interestThe authors state that they have no conflicts of interest at the time of drafting of the present manuscript.
Please cite this article as: Lima-Martínez MM, Paoli M, Vázquez-Cárdenas A, Magaña-Torres MT, Guevara O, Muñoz MC, et al. Frecuencia, aspectos clínicos y moleculares de la hipercolesterolemia familiar en una unidad de endocrinología de Ciudad Bolívar, Venezuela. Endocrinol Diabetes Nutr. 2017;64:432–439.
