




El tumor de células granulares (TCG) pertenece al grupo de tumores de la hipófisis posterior (clasificación de la WHO de 2017)1. Son tumores poco frecuentes, con la característica común de tener inmunohistoquímica positiva para el factor de transcripción tiroideo 1 (TTF-1), proteína S-100 y vimentina. La coexistencia con adenomas hipofisarios funcionantes es excepcional, con muy pocos casos descritos hasta la fecha2. Describimos el caso clínico de una mujer con un TCG de hipófisis posterior asociado a una enfermedad de Cushing.
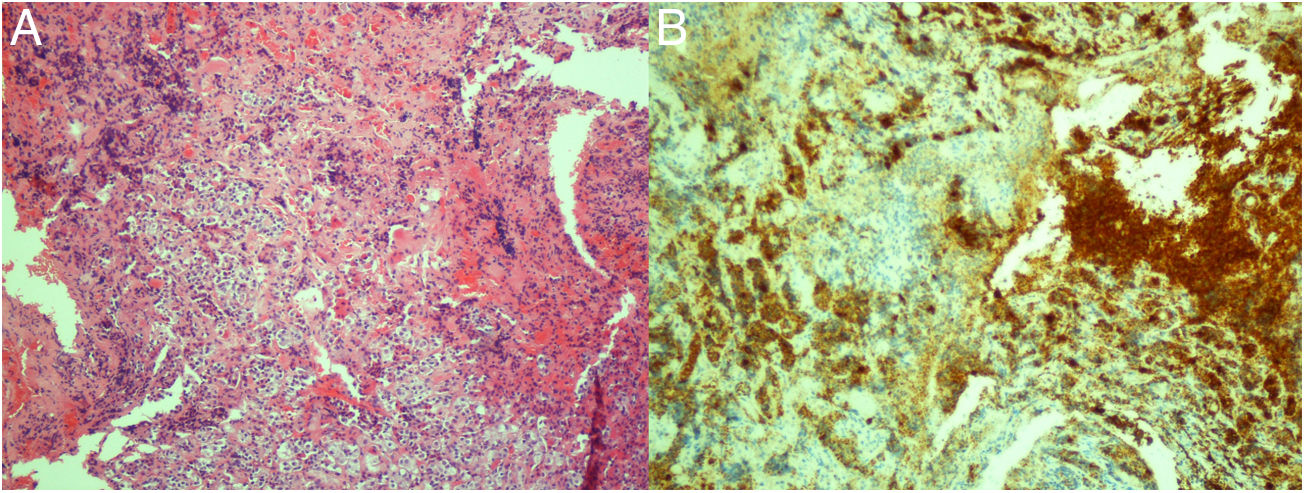
Mujer nacida en 1954 con historia de hipertensión arterial esencial, diabetes mellitus tipo 2 y meningioma frontal derecho. En 2014 se diagnosticó de síndrome de Cushing a raíz de cambios en el fenotipo facial, hipotrofia muscular, aumento del perímetro abdominal, hematomas espontáneos y fracturas costales. No presentaba estrías. Su perímetro abdominal era de 101cm, peso 60,9kg, talla 154cm y presión arterial 165/88mmHg. El estudio funcional bioquímico sugirió un hipercortisolismo endógeno de origen hipofisario [cortisol libre urinario (CLU) 318μg/g creatinina (Cr) (normal 10-100μg/g Cr)]. Sin embargo, la resonancia hipofisaria fue normal. Un cateterismo de senos petrosos confirmó el origen hipofisario del hipercortisolismo. Se decidió realizar una hemihipofisectomía endoscópica endonasal en mayo de 2016. Durante el acto quirúrgico se observó una hipófisis normal, pero con una zona posterolateral fibrosa sospechosa, que se resecó. El estudio anatomopatológico informó de la presencia de glándula hipofisaria normal en uno de los bloques. En el otro, se observó la presencia de un tumor fibrilar compuesto de células fusiformes, epiteloides y granulares, positivas para proteína S-100 y TTF-1, compatible con TCG. Se observaron en su interior nidos de células positivas para sinaptofisina y ACTH, concordantes con un corticotropinoma (fig. 1).
 Tinción de hematoxilina y eosina. B) Inmunohistoquímica para ACTH.")
La cirugía no fue curativa (CLU 311,6μg/g Cr a los 2 meses de la intervención), por lo que se decidió tratamiento con radioterapia estereotáxica fraccionada (dosis acumulada de 51Gy en 28 sesiones) y se inició ketoconazol adyuvante (800mg cada 24h) en julio de 2016. Pese a las dosis altas de ketoconazol no se consiguió normalizar las concentraciones de cortisol libre en orina, por lo que se sustituyó ketoconazol por pasireótida 20mg cada 28 días. El tratamiento con pasireótida normalizó las concentraciones de CLU (87μg/g Cr) y redujo significativamente las concentraciones circulantes de ACTH y de cortisol sérico, pero deterioró el control metabólico de la diabetes previa que presentaba la paciente. Tras 3 años de tratamiento con pasireótida y 4 años tras la radioterapia, la paciente presenta criterios bioquímicos de enfermedad controlada, con gran mejoría clínica. La dosis actual de pasireótida es de 20mg cada 6 semanas. La diabetes se controla adecuadamente con sitagliptina 100mg/día y dapagliflocina 10mg/día.
La coexistencia de adenomas hipofisarios funcionantes y de tumores de hipófisis posterior es excepcional. En una revisión publicada recientemente2, se describen 10 casos de tumores de hipófisis posterior asociados a hipercortisolismo, 7 de ellos con histología de pituicitomas y 3 con un TCG. Actualizando los datos reportados por dicha revisión, en el momento de la escritura de este caso clínico, existen 9 pituicitomas asociados a síndrome de Cushing y tan solo 2 TCG, el reportado por Zhang el at.3 y el actual caso clínico, ya mencionado previamente4. La discrepancia en la revisión de 2019 con los datos mencionados se encuentra en los 3 casos clínicos reportados por Feng et al.5, siendo en realidad 2 pituicitomas (uno asociado a hipercortisolismo y otro a acromegalia) y solo un TCG, asociado a acromegalia. El noveno caso de pituicitoma asociado a enfermedad de Cushing se ha publicado recientemente6. Hasta la fecha, hay descritos 5 casos de tumores de hipófisis posterior asociados a acromegalia, 2 de ellos con histología de TCG2. Guerrero et al.2 teorizan que la existencia de un tumor de hipófisis posterior podría producir sustancias (citoquinas, factores de crecimiento) que estimulasen la proliferación de las células de la adenohipófisis, favoreciendo la aparición de tumores adenohipofisarios funcionantes.
Los hallazgos clínicos de los tumores neurohipofisarios son indistinguibles de los macroadenomas hipofisarios no funcionantes, siendo más frecuente su presentación como alteraciones visuales (64%) o cefalea (33%)1. Habitualmente, se observa una lesión ocupante de espacio en las pruebas de imagen, y el diagnóstico definitivo se obtiene con el resultado de la anatomía patológica. Los hallazgos histológicos son superponibles a los descritos en el caso clínico, pero como característica diferencial frente a los pituicitomas y oncocitomas fusocelulares, los TCG son positivos en la tinción con ácido peryódico de Schiff (PAS)1.
Existe un acuerdo de que el tratamiento de este tipo de tumores debe ser quirúrgico, a ser posible por vía transesfenoidal. El pronóstico es dependiente de la resección completa4. Por su localización anatómica y su mayor tendencia al sangrado (son estructuras más vascularizadas), la cirugía es más compleja que la realizada en adenomas hipofisarios4,7.
El presente caso describe un TCG con una infiltración de células corticotropas, responsable de la hiperproducción de ACTH, sin evidencia de masa selar. Casualmente, el otro caso descrito de síndrome de Cushing y TCG3 tampoco visualizaba masa tumoral en la resonancia magnética, sin embargo, se logró el control bioquímico tras la intervención. En el presente caso, fue necesario el uso de radioterapia como tratamiento adyuvante para el control del hipercortisolismo a largo plazo. Como tratamiento médico, se utilizó ketoconazol, sin respuesta, por lo que se decidió cambiar por pasireótida.
La pasireótida es el primer agente con efecto directo sobre la secreción hormonal hipofisaria aprobado en ficha técnica para el tratamiento de la enfermedad de Cushing. Presenta una alta afinidad por el receptor de somatostatina tipo 5 (SSTR5), el cual se expresa fuertemente en los corticotropinomas, habiendo demostrado normalización y reducción del hipercortisolismo en sus estudios pivotales8. En «vida real»9 se describe un 68% de normalización bioquímica del hipercortisolismo en enfermedad de Cushing resistente o recurrente (tratada previamente con cirugía, radioterapia o tratamiento médico). Esto sugiere que deben existir una o varias características diferenciales en el subgrupo de tumores respondedores a pasireótida, quizá una mayor expresión de SSTR5. Es posible que la patogenia sugerida para la asociación de 2 tumores de glándulas contiguas pero diferentes pueda explicar la excelente respuesta de la hipersecreción de ACTH al tratamiento con pasireótida vista en el caso descrito. Obviamente sería necesario disponer de una serie mucho más grande para llegar a una conclusión. Desafortunadamente, no disponemos de datos moleculares de la expresión de receptores de somatostatina en este tumor, por lo que no es posible establecer su relación con la respuesta a pasireótida.
Se presenta el caso de una paciente con un TCG con nichos de células corticotropas en su interior responsables de una enfermedad de Cushing con excelente respuesta al tratamiento con pasireótida.
Aspectos éticosEl consentimiento informado es obtenido de todos los pacientes intervenidos de patología hipofisaria en nuestro centro, donde aceptan la inclusión de sus muestras en el biobanco y el uso de sus datos clínicos anonimizados en la divulgación científica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.
