


Ganglioneuroma is a benign neoplasm derived from the neural crest consisting of mature ganglionic cells and Schwann cells.1 The posterior mediastinum is its most common location, followed by the retroperitoneum. Only a small proportion of ganglioneuromas arise from the adrenal medulla (15–30%).1
The routine use of imaging tests has increased the frequency of diagnosis of adrenal incidentalomas. Most of these lesions are nonfunctional benign adenomas. Other less common lesions include cortisol-secretin adenoma, metastases, adrenal carcinoma, and myelolipoma. Lesions such as cysts, inflammatory or infectious lesions, bilateral adrenal bleeding or, as in the case reported here, ganglioneuroma are less frequently found.2
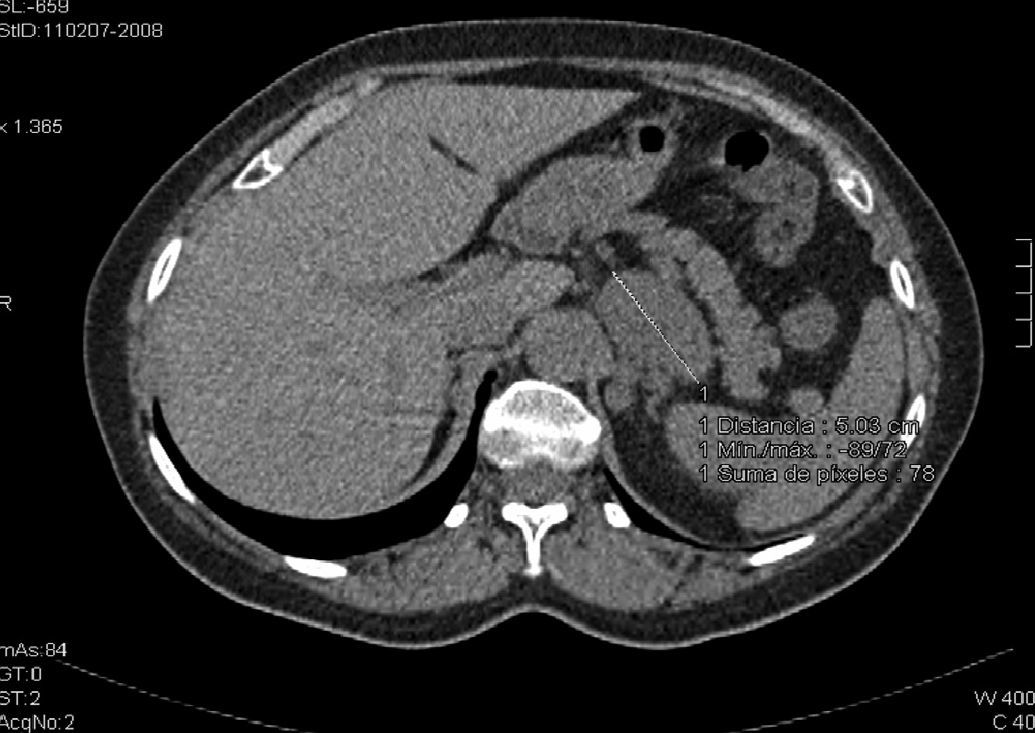
Our clinical case was a 61-year-old with a history of pemphigus vulgaris in 1998 who required high-dose corticosteroid therapy. She reported no known allergic reactions to drugs, and had no history of high blood pressure, diabetes mellitus, or dyslipidemia. A computed tomography (CT) scan of the chest performed for chronic cough revealed a left adrenal mass 61mm×47mm×37mm in size with lobulated contours and attenuation values of 20–30 Hounsfield units (HUs) in basal slices (Fig. 1; oral and intravenous contrast was not used because the radiologist suspected pheochromocytoma based on basal images). The patient was referred to the endocrinology department to work-up a left adrenal incidentaloma. The patient reported pain at rest in the left lumbar region. Upon specific questioning, she reported no constitutional symptoms or evidence of hypercortisolism (central obesity, striae, muscle weakness, bruising, mood changes, hirsutism, acne), hyperandrogenism, or hyperaldosteronism (HBP, muscle weakness, nocturia, urinary frequency, polydipsia), or crises suggesting pheochromocytoma.

Physical examination revealed the following data: weight, 57.5kg; height, 150cm; BMI, 25.6kg/m2; waist circumference, 92cm; supine BP, 155/95mmHg, and standing BP, 145/90mmHg. The thyroid gland was normal, and there were no neurofibromas, lesions suggesting cutaneous amyloidosis, or clinical evidence of hypercortisolism or hyperandrogenism. No masses were found on abdominal palpation. The results of complete blood count and general chemistry tests were normal. The results of tests in 24-h urine were as follows: creatinine, 1.0g/24h (0.6–2.0); urine output, 1800mL (1000.0–1800.0); cortisol, 212.40μg/24h (42.0–218.0). Results in a second 24-h urine sample were as follows: epinephrine, <4μg/24h (0.0–18.0); norepinephrine, 31μg/24h (0.0–80.0); dopamine, 166μg/24h (0.0–400.0); fractionated metanephrine, 167μg/24h (74.0–297.0); fractionated normetanephrine, 164μg/24h (105.0–354.0); total metanephrines, 331μg/24h (170.0–700.0). Repeat urinary free cortisol and metanephrine tests were normal in the absence of drugs with the potential to interfere with their measurement. The results of plasma hormone tests were as follows: testosterone, 0.28ng/mL (0.15–0.71); 17-OH-progesterone, 0.2ng/mL (0.0–0.0); androstenedione, 0.5ng/mL (0.9–3.0); DHEAS, 42.7μg/dL (59.0–296.0); ACTH, 13.8pg/mL (0.0–46.0); cortisol, 11.8μg/dL (6.0–28.0); epinephrine, <20pg/mL; norepinephrine, 392pg/mL, dopamine, <20pg/mL; renin, <0.10ng/mL/h (1.9–6.0); aldosterone, 197pg/mL (35.0–275.0).
Abdominal magnetic resonance imaging (MRI) with T1- (in- and out-of-phase) and T2-weighted axial slices detected a left adrenal mass 56mm in anteroposterior diameter, 56mm in craniocaudal diameter, and 35mm in cross-sectional diameter which showed an intermediate signal in T1-weighted sequences, with no signal hyperintensity foci or signal loss in out-of-phase sequences, which excluded lipid contents. Low signal predominated in T2-weighted sequences, with signal hyperintensity in the upper and middle zones. Low contrast uptake was seen in the rest of the adrenal mass, with an almost avascular central area. Radiographically, this suggested a chromaffin cell tumor, although adrenal metastasis or adrenocortical carcinoma could not be ruled out. No adenopathies in the rest of the retroperitoneum or liver lesions suggesting metastasis were identified.
Carcinoma was suspected based on size and radiographic characteristics, and surgical resection was decided upon. Transperitoneal laparoscopic surgery was performed because of the extensive experience of the surgeon with this approach. The operating time was 60min, and there were no surgical complications. A pathological study disclosed an adrenal mass of lobulated appearance 6cm×4.5cm×2.5cm in size. At microscopic examination, the mass was found to consist of bundles of fusiform cells with slightly eosinophilic cytoplasms and nuclei with no cell atypia with morphology of Schwann cells, interspersed with neurons without signs of cell atypia, but no mature chromaffin tumor cells were seen. At immunohistochemistry, the tumor was positive for S100, neuron-specific enolase, synaptophysin, and CD57, which supported a diagnosis of adrenal ganglioneuroma. Abdominal CT scans performed 3 and 12 months after surgery showed no recurrence of the initial lesion or evidence of other pancreatic or thyroid tumors.
Ganglioneuroma is a benign tumor derived from the neural crest occurring in the paravertebral sympathetic ganglia or the adrenal medulla. The tumor consists of mature ganglion cells and Schwann cells on nerve fibers.1 Ganglioneuroma most commonly occurs in children and young adults.1,3 The tumor similarly affects males and females (1.13:1) and its most common location is the retroperitoneum (35–52%), followed by the mediastinum (39–43%) and the cervical region (8–9%).1
These masses are usually asymptomatic, and the clinical signs are usually non-specific and vary depending on the location. They are usually detected in imaging tests performed for other unrelated reasons.4 Tumors occasionally secrete catecholamines and cause adrenergic clinical signs similar to those of mature chromaffin cell tumors,5,6 particularly if the tumor consists of paraganglia precursor cells (ganglioneuroma) and mature chromaffin cells (pheochromocytoma), in which case it is called composite pheochromocytoma.7 Other less common presentation forms include hirsutism due to testosterone hyperproduction,8 glomerulonephritis related to the tumor that resolves upon surgical resection,9 and diarrhea caused by the production of vasoactive intestinal peptide.6
On gross examination, the tumors are well circumscribed and encapsulated. They are of variable size, 8cm on average, but tumors weighing up to 5kg have been reported.10
In a radiographic study of an adrenal incidentaloma, malignancy predictors should be assessed carefully before deciding on a conservative approach: by contrast CT, in lesions with basal HU values >10, absolute contrast washout <50% has 100% sensitivity and specificity for detecting malignant lesions and chromaffin cell tumors. Data suggesting malignancy, such as size >4cm, necrosis, and intralesional bleeding, should also be taken into consideration.11 In recent years, informative studies have been published concerning the discriminant role of fluorodeoxyglucose positron emission tomography (PET-TC) in the preoperative diagnosis of malignant adrenocortical lesions. In a prospective cohort of 77 patients with histologically documented adrenocortical lesions, an adrenal/liver SUVmax ratio in PET-TC showed 100% sensitivity and 88% specificity for differentiating adenomas from adrenocortical carcinomas.12 There are however no studies supporting the value of PET-TC with fluorodeoxyglucose to distinguish ganglioneuromas from other adrenal lesions, all the more so because ganglioneuroma is a benign tumor, and fluorodeoxyglucose is attributed a specific value as a marker of dedifferentiation and progression in tumor disease.13
Radiographically, ganglioneuromas appear within the spectrum of potentially malignant lesions: in CT attenuation is usually lower than 40HUs, and in MRI they have a low signal intensity in T1 and higher and heterogeneous intensity in T2. It is therefore essential to distinguish the differential traits of ganglioneuromas.14 Maweja et al. recommend that ganglioneuroma be suspected when lesions have an attenuation in CT ranging from 10 to 40HUs with no hormone hyperproduction, vascular invasion, or fine calcifications. In MRI, ganglioneuromas are hypointense in T1 and hyperintense in T2 with a heterogeneous pattern,15 which contrasts with the usually homogeneous hyperintensity of pheochromocytomas in T2, provided they have no distinguishable necrotic or hemorrhagic areas in T1 sequences.
Prognosis is excellent. Treatment is complete surgical resection without the need for chemotherapy or radiotherapy because of the benign nature of the lesion.
The case reported was unique in different aspects. On the one hand, the patient was older than in the other cases reported, which emphasizes the need for the diagnostic approach to adrenal masses to be based on their radiographic and biochemical characteristics, while clinical data help orient the suspicion but are not confirmatory in any case. In addition, PET-CT is increasingly used in the study of adrenal incidentalomas, and in this case it would have been very helpful, but the patient declined the test to avoid delays in surgery.
As regards laparoscopic indication for malignant adrenal gland disease, we think that a distinction should be made between primary lesions (adrenocortical adenoma) and metastases from other primary tumors (lung, colorectal, melanoma, kidney, breast). Adrenal primary carcinoma is an uncommon tumor, with an incidence of 1–2 cases/1,000,000 inhabitants/year. It is an aggressive tumor with a 5-year survival rate ranging from 11% to 38% and which should be treated with complete surgical resection. Initial laparoscopic management of this type of tumor was an absolute failure, with five cases of local and portal recurrence and peritoneal carcinomatosis reported in the 1997–1999 period.16–20 This led to the suspicion that laparoscopy facilitated the dissemination of tumor cells, probably due to pneumoperitoneum. Subsequent series, with a still small sample because of the infrequency of the disease, achieved better oncological results than those initially reported.18,21–23
A meta-analysis published in 200524 of 420 open adrenalectomies for malignant disease found a local and peritoneal recurrence rate very similar to that reported in the laparoscopic series (30% local, 67% distant, and 14% peritoneal recurrences). It is, of course, mandatory to follow the basic rules of oncological laparoscopic surgery: the use of a specimen retrieval bag, the avoidance of tumor specimen rupture, and complete resection of the mass with a safety margin. Patient selection is also important. It appears reasonable to avoid large tumors where there is radiographic suspicion of an infiltration of adjacent structures. In these cases, the possibility of conversion to open surgery, the risk of local recurrence, peritoneal dissemination, or metastases in portals is greater.25–27
Finally, the association of adrenal ganglioneuroma with hereditary multiple endocrine neoplasia syndromes is exceptional, but a study of RET or VHL should be considered in patients with adrenal ganglioneuromas when they are combined with cutaneous stigmata or other tumors more classically associated with such syndromes (pheochromocytoma, medullary thyroid carcinoma, or pancreatic neuroendocrine tumors), or in subjects with bilateral adrenal ganglioneuromas, particularly if they occur in the first decades of life.28–31 Our patient had no such stigmata, and a genetic analysis of RET or VHL was therefore not requested. However, such considerations regarding the genetic study are the same for composite tumors (ganglioneuroma–pheochromocytoma or ganglioneuroma–paraganglioma) as for isolated chromaffin cell tumors. Careful examination of the histological specimen is therefore particularly important before a diagnosis of ganglioneuroma is made.
In conclusion, in work-up of adrenal incidentalomas, other less common etiologies such as ganglioneuroma should be considered, and the latter should be suspected in masses with a malignant radiographic appearance without associated hormone hyperproduction. The reported case also shows the importance of interdisciplinary collaboration between surgeons, radiologists, and endocrinologists experienced in adrenal conditions to optimize clinical management.
Please cite this article as: Olivar J, Fernández A, Aguilera A, Diaz P, Martín V, Lahera M. Ganglioneuroma adrenal: dilema clínico-quirúrgico acerca de un hallazgo fortuito. Endocrinol Nutr. 2013;60:e37–e40.
