


Type 1 diabetes mellitus is associated with other autoimmune diseases, most commonly with lymphocytic thyroiditis or celiac disease. Some cases of limbic encephalitis associated with type 1 diabetes, thyroiditis or other autoimmune conditions have been reported in the past decade, even in children.1–5 Limbic encephalitis is an autoimmune inflammatory process involving the hippocampus and amygdala which until a few years ago was considered to be of paraneoplastic origin. Patients have in blood and/or CSF antibodies (Ab) against the neuronal surface or intracellular antigens, such as anti-GAD Ab. GAD, selectively expressed in neurons and pancreatic cells, is the enzyme that limits the synthesis rate of GABA (aminobutyric acid), the main inhibitory neurotransmitter that modulates and synchronizes neuronal activity in CNS. These Ab inhibit GAD activity, possibly mediated by cytotoxic T cells, and reduce GABA synthesis or exocytosis. They cause neuropsychiatric changes such as hallucinations, refractory temporal epilepsy, memory dysfunction and cognitive impairment, which may be reversed with immunomodulatory treatment.4–6
A patient has “limbic syndrome” if he/she meets more than one of the following criteria: recent memory impairment, temporal lobe crisis, or psychiatric abnormalities, and also one of the following: neuropathology (chronic medial temporal encephalitis); a tumor diagnosed within five years of the appearance of neurological symptoms and signs; onconeuronal antibodies or VGKC, NMDAR, GAD Ab; the finding in brain MRI of an unexplained increase in the FLAIR/T2 medial temporal signal.6–9
The case of a girl with the onset of type 1 diabetes mellitus at four years of age and who experienced cognitive impairment and behavioral disorder at seven years of age is reported below. She had no family or personal history of interest. Psychomotor development had been normal until 7 years of age, but progressive behavioral disorders (aggressiveness, disobedience), memory loss, poor school performance, irrational fear, anxiety, auditory hallucinations, sleep disturbance, and absence seizures, subsequently occurred. The latter increased in severity and frequency, showing signs of complex partial seizures. At 10 years of age, the patient had multiple daily episodes consisting of facial clonus, ocular version, hypertonia, and generalized tonic–clonic seizures. Cognitive impairment and school performance simultaneously worsened, particularly as regards executive dysfunction and memory loss. She also showed poor emotional and impulse control, severe behavioral disorders and aggressiveness (she hit her classmates, took off her clothes in class), and required a special education school.
Despite intensive treatment of diabetes with insulin detemir and lispro following a basal bolus regimen and diet, control was poor because of the multiple daily crises causing hyperglycemia and hypoglycemia; HbA1c 7.8%, TSH 3.00, FT4 0.79, negative microalbuminuria, negative thyroid Ab. Negative transglutaminase IgA Ab. The patient also had early puberty, for which she was treated with triptorelin.
Because of the refractoriness of her epileptic seizures, the patient was referred at the age of 12 to a tertiary hospital where potential surgical treatment for epilepsy was assessed.
- -
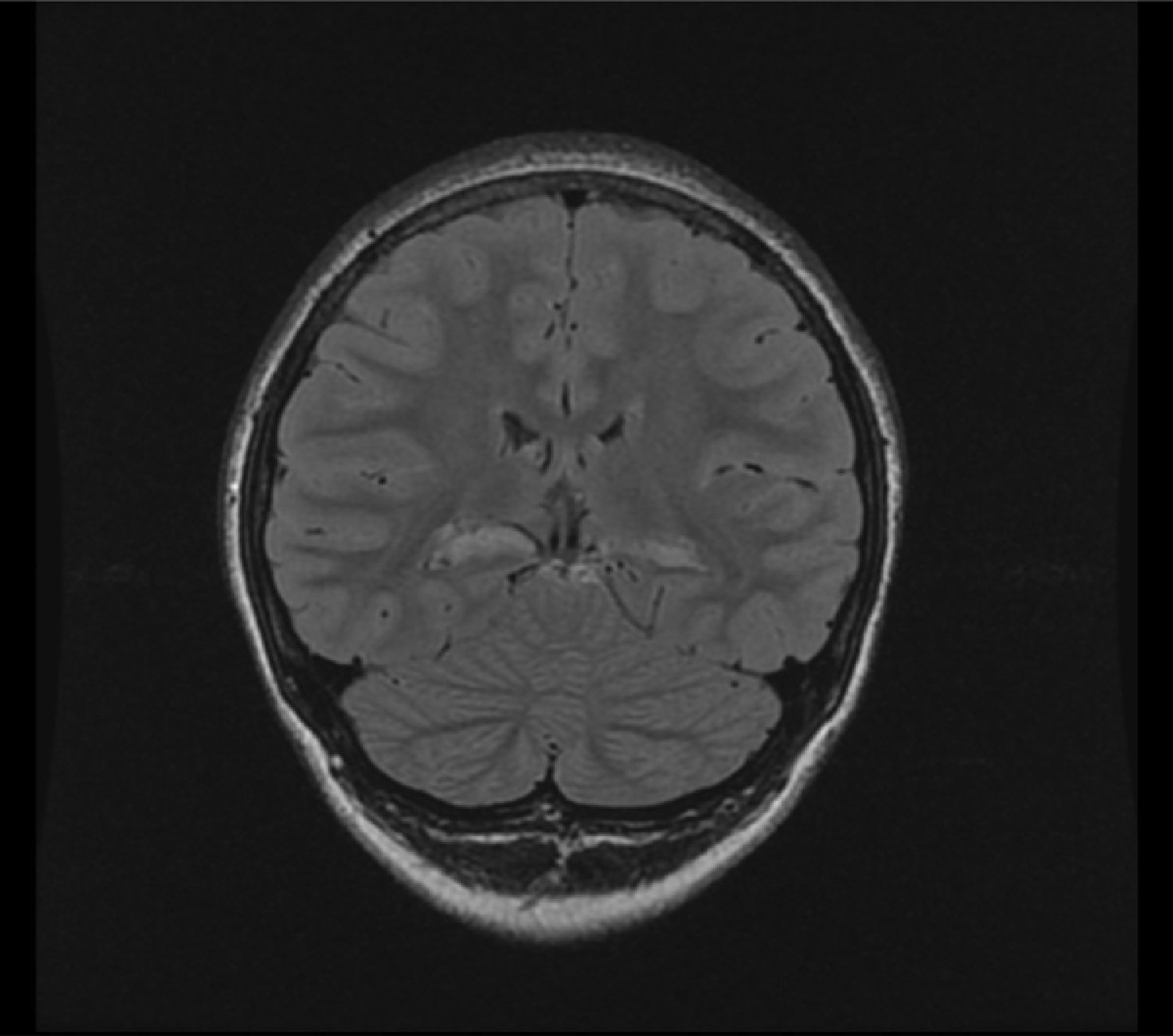
3T MRI performed there showed atrophy of both hippocampi, with increased T2 and FLAIR signal, suggesting bilateral medial temporal sclerosis. There was a greater decrease in left hippocampal head volume, and more severe signal changes in the right side (Fig. 1).
- -
PET/CT: bilateral temporal hypometabolism, more marked and extensive in the right temporal lobe, affecting the mesial region, temporal pole, and neocortex.
- -
Video EEG: intercritical abnormalities: right and left temporal epileptiform abnormalities: 26 right temporal crises and 4 generalized tonic–clonic seizures.
- -
Neuropsychological assessment revealed cognitive/evolutionary impairment in all domains, an intelligence quotient previously 69, currently 51, impaired memory, especially verbal, as well as executive dysfunction, difficulties in emotional and impulse control, severe behavioral disorder, dissocial behavior.

She was diagnosed with refractory epilepsy with bilateral medial temporal sclerosis, mainly on the right side, refractory to drug treatment. A right temporal lobectomy with right amygdalohippocampectomy was therefore performed.
Three months after surgery the seizures had improved, but the schizophrenia-like psychotic picture had worsened, which led to the etiology being reassessed:
- -
Neuronophagia was seen in a surgical biopsy, and with CD3 staining for lymphocytes, “microglial nodules” of lymphocytes appeared, both consistent with encephalitis. Autoimmune encephalitis antibodies were tested with negative results (NMDA, AMPA,GABA, mGLuR1, mGLUR5, VGKC, and GAD). Oligoclonal bands in CSF: positive. Abdominal ultrasonography: normal.
- -
Based on histological findings consistent with encephalitis, autoimmune endocrine disease (type 1 diabetes), and oligoclonal bands in CSF, “autoimmune encephalitis” was suspected and immunotherapy was started. Immunomodulatory treatment was started with megadoses of methylprednisolone and monthly cycles of immunoglobulin IV (2g/cycle). Marked improvement in school performance (memory, language, and calculations) and behavior was seen from the third cycle, and the seizures disappeared.
- -
Since the age of 15 years, after 3 years of immunotherapy, the patient has required no treatment and has had no other diseases. She has both good family and school integration, with no behavioral changes. Her cognitive evolution has been favorable, with improvements in school performance, language, attention, and memory. The auditory hallucinations have disappeared. She has no seizures, but takes oxcarbamazepine due to sporadic EEG abnormalities. Diabetes control has improved with the same regimen: feeding by portions and basal bolus insulin detemir and lispro.
- -
Normal physical examination, weight 51kg (−0.61 SD); height 151cm (−1.78 SD); Tanner V; HbA1C 6.7%; TSH 3.1 mcIU/mL; FT4 0.90ng/dL; cholesterol 176mg/dL; ACTH 43.8pg/mL; cortisol 22μg/dL, and negative transglutaminidase Ab. Negative microalbuminuria and negative thyroid Ab.
This case is interesting because of the recent identification of autoimmune origin in the limbic system and its rarity among the pediatric population.3,4 It has been reported that GAD Ab are associated with a greater risk of neurological impairment because of the role of GAD in GABA synthesis (especially with high levels).5,10 Intrathecal Ab synthesis may persist for a long time, causing the clinical condition five years after its detection in blood,6 and not always being detected.4 These conditions occur at younger ages than other types of autoimmune encephalitis, and appear to respond better to treatment than those caused by other Abs against intracellular antigens.6 It is also interesting due to the difficulties in making the diagnosis, which was only arrived at after right temporal lobectomy indicated for refractory epilepsy with neurocognitive impairment. Just because of its rarity and novelty, an autoimmune origin was only suspected after histological findings consistent with encephalitis, and was confirmed when work-up was completed. The clinical course of this girl was noteworthy because, after years of seizures and progressive neurological, cognitive, and behavioral impairment, with no improvement after neurosurgery, she showed an excellent response to immunoglobulins, with no relapse or side effects. We think that this case may help us to identify and diagnose similar cases in the future, to avoid neurosurgery, and to promote the earlier use of immunoglobulins based on their efficacy.4,6–8
Please cite this article as: Temboury Molina MC, Ruiz-Falco Rojas ML, Palmi Cortés I, Villamor Martín R. Encefalitis límbica autoinmune en una niña con diabetes tipo 1. Hallazgos clínicos y evolución. Endocrinol Nutr. 2016;63:308–310.
