


Neurofibromatosis type 1 (NF-1) or von Recklinghausen's disease is one of the syndromes traditionally associated with pheochromocytoma. NF-1 is a multisystemic disease that mainly affects the skin and nervous system, of dominant autosomal inheritance and highly variable clinical expression even in members of the same family. In up to 50% of patients, de novo mutations occur and there is no family history.1
The neurofibromatosis gene is located in chromosome 17 and encodes for neurofibromin, a protein involved in the regulation of p21 ras which acts as a tumor suppressor. Loss of protein function therefore allows for uncontrolled cell proliferation and the occurrence of various tumors.2
The diagnosis of NF-1 is clinical and is established when at least two of the following criteria are found3,1 six or more cafe-au-lait spots,2 two or more skin neurofibromas or a plexiform neurofibroma,3 axillary freckling,4 Lisch nodules,5 optic glioma,6 bone dysplasia, and7 a first-degree relative with NF-1. Ninety-seven percent of patients meet the criteria at eight years and 100% at 20 years. Genetic diagnosis is possible in more than 85% of patients,1 but is technically complex and does not predict for the occurrence of complications because there is usually no phenotype–genotype correlation. Thus, routine genetic diagnosis is not indicated.4
One of the multiple manifestations of NF-1 is pheochromocytoma, occurring in 1–5% of patients.5 Familial pheochromocytoma in the setting of neurofibroma is rare, and few cases have been reported.6 Two cases of pheochromocytoma occurring in a mother and her daughter with NF-1 are reported, and the most relevant aspects of this association are reviewed.
Case 1: A female patient with no relevant personal or family history, except for hypertension in her mother, who died from stroke at 54 years of age. Her father and five siblings were healthy. She was diagnosed with NF-1 at 36 years of age based on an inguinal tumor biopsy consistent with neurofibroma. A physical examination revealed more than six cafe-au-lait spots, small tumors in the fingers of both hands, and Lisch nodules in the ophthalmological evaluation. After diagnosis, her four children were evaluated and were all found to meet NF-1 criteria.
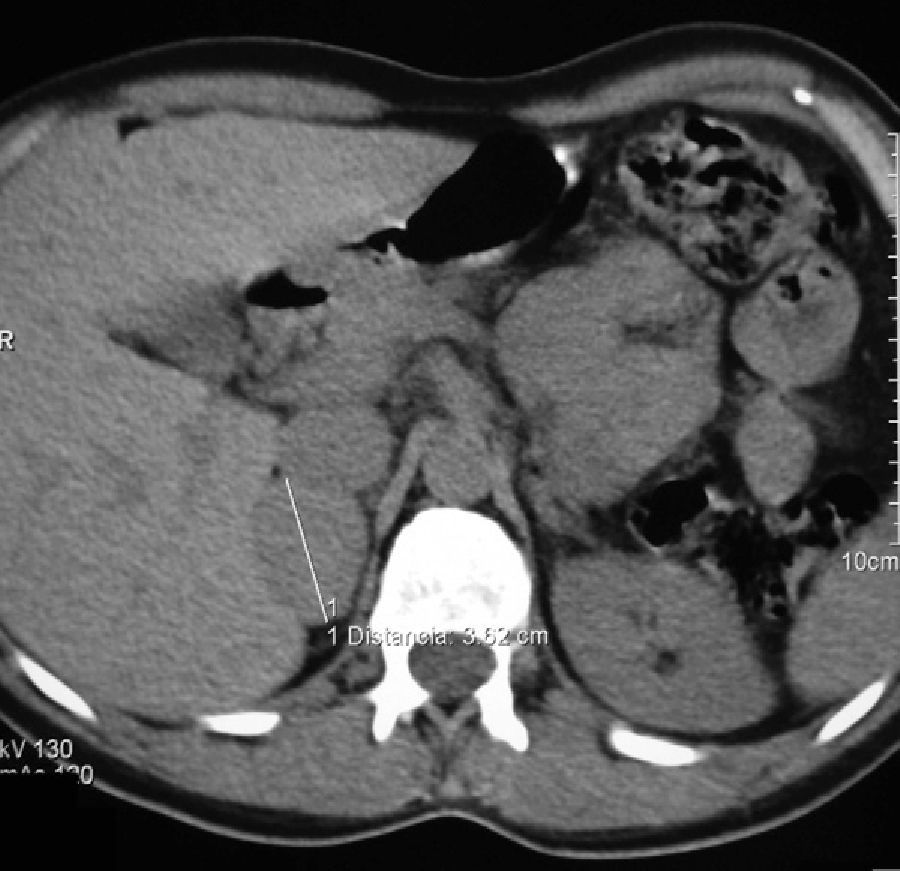
At the age of 40, she underwent work-up for dyspepsia and diarrhea, and an abdominal ultrasound examination revealed a right adrenal nodule. The patient reported sweating and heat intolerance over the previous two months, and her BP at the consultation was 158/92mmHg. There were no changes in routine laboratory tests. Urinary catecholamine and metanephrine tests were performed, with the following results: epinephrine, 117mcg/24h (NR: 0.5–20mcg/24h); norepinephrine, 424mcg/24h (NR: 14–80mcg/24h); metanephrine, 1386mcg/24h (NR: 86–320mcg/24h); and normetanephrine, 1720mcg/24h (NR: 129–400mcg/24h). An abdominal CT scan confirmed the presence of a homogeneous right adrenal nodule, 3.5cm in size, of 44 HU (Fig. 1). Meta-I-benzylguanidine (MIBG) scintigraphy showed uptake in the right adrenal gland. Laparoscopic surgery was performed after pharmacological alpha blockade with phenoxybenzamine 10mg/day. The pathological study was consistent with encapsulated pheochromocytoma.
.")
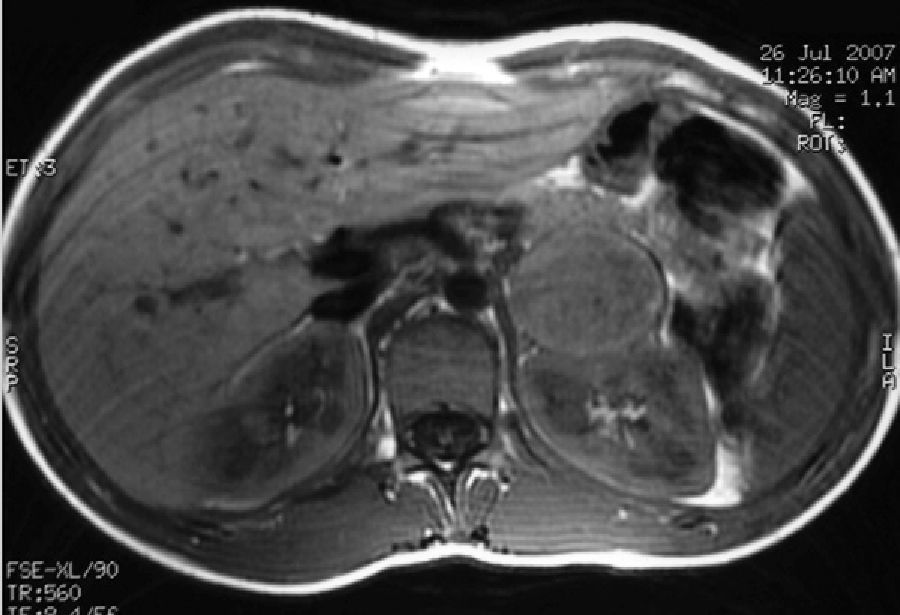
Case 2: A female patient diagnosed with NF-1 at nine years of age after the diagnosis of her mother (case 1). At 15 years of age she reported episodes of headache and palpitations. Physical examination found more than six cafe-au-lait spots and BP values of 154–83mmHg. Laboratory tests revealed basal blood glucose of 128mg/dL with no other changes, and a CT scan of the head and cardiological evaluation were normal. Urinary catecholamine and metanephrine tests were performed, with the following results: epinephrine, 150mcg/24h (NR: 0.5–20mcg/24h); norepinephrine, 713mcg/24h (NR: 14–80mcg/24h); metanephrine, 1757mcg/24h (NR: 86–320mcg/24h); and normetanephrine, 3967mcg/24h (NR: 129–400mcg/24h). MRI showed a nodule in the left adrenal gland, 4cm×4cm×3.8cm in size, hyperintense in T2, heterogeneous, and with necrotic areas (Fig. 2). MIBG scintigraphy showed left adrenal hyperfixation. Laparoscopic surgery was performed after treatment with phenoxibenzamine 20mg/12h, and a pathological examination confirmed the diagnosis of pheochromocytoma.
.")
Both patients have remained asymptomatic and normotensive since surgery, with annual catecholamine and metanephrine tests showing normal values. Catecholamine and metanephrine tests requested in the remaining children of case 1 gave normal results.
Pheochromocytoma occurs in 1–5% of patients with NF-1.5 Unlike pheochromocytoma associated with other genetic syndromes, its clinical characteristics are similar to those of sporadic pheochromocytoma.7 Most of these tumors occur in adults with a mean age of 42 years (range, 1.5–75 years).8 Patients usually have high blood pressure (HBP) and/or consistent clinical signs, but up to 22% may be asymptomatic and normotensive.8 Unifocal adrenal tumors are most common, but 10–20% are multifocal7–10 and 6% extraadrenal.5,8 Up to 12% are malignant.7,8 The reported cases were unifocal adrenal with associated clinical signs and HBP. In case 1,the diagnosis was made after an adrenal mass was incidentally found, but the patient had consistent clinical signs and HBP. NF-1 is usually diagnosed in childhood,1 and the finding of apparently sporadic pheochromocytomas is therefore unlikely. There are however mild phenotypes that may have been overlooked, and signs of NF-1 should therefore be sought in apparently sporadic pheochromocytomas.
The range of mutations detected in patients with NF-1 and pheochromocytoma is very wide, and no specific mutations favoring its occurrence or conditioning its presentation or malignancy are known. A study by Neuman et al. attempted to describe the mutational spectrum in 37 patients with pheochromocytoma and NF-1. The authors found 36 different mutations distributed throughout the gene with no significant correlations with specific hot spots.5
Biochemical and imaging diagnostic tests do not differ from those used in sporadic pheochromocytoma. Pheochromocytoma associated with NF-1 predominately secretes epinephrine and induces high metanephrine and normetanephrine levels in plasma and urine.9
Surgery, preferably by laparoscopy, after preparation with alpha-blockers is the treatment of choice. Subsequent follow-up is the same as for sporadic pheochromocytoma. Annual measurement of catecholamines and metanephrines is recommended. The results of these tests in our patients have been normal.
Because of the low frequency of pheochromocytoma in patients with NF-1, routine biochemical screening is not indicated.8 In children and adults with mild, asymptomatic phenotypes, the clinical guidelines recommend at least annual monitoring for typical symptoms and BP measurement.1–3 In patients with clinical signs or HBP, catecholamines and metanephrines should be tested in blood and/or urine. HBP is common in patients with NF-1, and although its most common causes are hypertension in adults and renal artery stenosis in children, the presence of pheochromocytoma should always be ruled out.2 This condition should also be ruled out in patients who plan to become pregnant or are scheduled to undergo any medical procedure.8
However, routine screening could increase the prevalence of pheochromocytoma up to 14.6%, as reported in a recent study,10 because a proportion of patients are asymptomatic and normotensive.
There are no specific recommendations for the management of relatives of patients with pheochromocytoma associated with NF-1. After the diagnosis of case 2, urinary catecholamines and metanephrines were measured in all other siblings affected with normal results. Ogawa et al. also performed these tests in a similar case of familial pheochromocytoma.6 Since with the same mutation there is less intrafamilial than interfamilial clinical variability, when pheochromocytoma is diagnosed in two members of a family with NF-1, biochemical screening of all other affected relatives would appear to be indicated.
Please cite this article as: Ollero García-Agulló D, Iriarte Beroiz A, Rojo Alvaro J, Munárriz P, Forga Llenas L. Feocromocitoma familiar asociado a neurofibromatosis tipo 1. Endocrinol Nutr. 2013;60:421–422.
