



Paragangliomas (PGLs) are uncommon neuroendocrine tumors (prevalence, 1/1700) derived from neural crest cell lines and occurring in adrenal medulla (pheochromocytoma [PHEO]), chemoreceptors, and sympathetic and parasympathetic ganglia.1 Clinical signs and symptoms depend on location, secretory profile, and malignant potential. Approximately 25% of PHEOs and PGLs are familial in origin and are part of different syndromes such as von Hippel-Lindau (VHL), multiple endocrine neoplasia type 2 (MEN2), neurofibromatosis type 1 (NF1), and the PGL/PHEO syndrome due to germline mutations in succinyl dehydrogenase enzyme (SDH),2 a enzyme involved in electron transfer and Krebs cycle and expressing a wide phenotypic heterogeneity.3
We report the case of a patient with paraganglioma/pheochromocytoma syndrome diagnosed 25 years after the occurrence of the first signs of the disease.
A 65-year-old male was admitted for constipation and abdominal pain over the previous 15 days and reported constitutional symptoms for 6 months. An endoscopy showed a stenosing mass in the rectosigmoid junction which required the placement of a Wallflex stent. Biopsy could not be performed.
Patient clinical history included type 2 diabetes mellitus, high blood pressure, and chronic obstructive pulmonary disease (COPD). He had been diagnosed in 1985 with bilateral carotid paragangliomas, treated by surgical resection, and a right tympanic paraganglioma. During the course of the disease, the patient had experienced two relapses that had been treated with radiotherapy and tomoradiosurgery. The father of the patient had died of cervical tumors, and his 30-year-old daughter had been diagnosed with a tympanic paraganglioma at 15 years of age and, more recently, with a carotid paraganglioma.
Laboratory test results included a blood glucose level of 169mg/dL and a carcinoembryonic antigen (CEA) level of 10.7ng/mL (0–5ng/mL). A CT scan of the abdomen and pelvis revealed a 4.3-cm necrotic mass in the interaortocaval retroperitoneum, a 2.6-cm left adrenal mass, a 1.5-cm right adrenal mass, and a mass in the rectosigmoid junction.
The patient reported headache, sweating, palpitations, tremor, and nervousness virtually daily over a period of several years, which had been attributed to an anxiety-depression syndrome and for which he was receiving long-term treatment with lorazepam. He was found to have an impaired general condition, dysphagia, right recurrent nerve palsy, a weight of 58kg, and BP levels of 105/58mmHg. The patient subsequently experienced paroxysmal hypertension. No striae, neurofibroma, Cushingoid habitus, cafe au lait spots, or Lisch nodules were found.
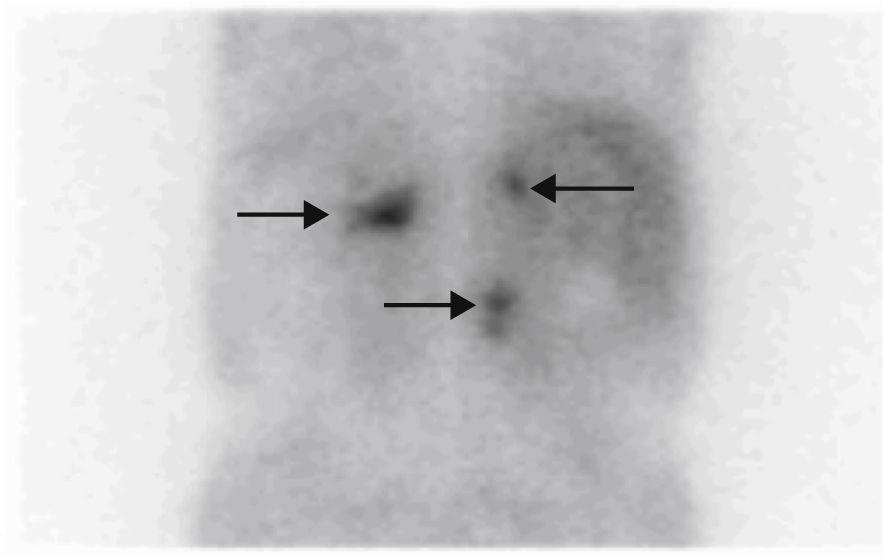
Based on his family history, head and neck paragangliomas, clinical signs and the symptoms of the patient, and new radiographic findings, the first diagnostic possibility considered was that the adrenal tumors were bilateral functioning pheochromocytoma and that the retroperitoneal tumor was a new abdominal paraganglioma, thus encompassing the whole clinical tumoral spectrum within the same condition, an SDH-related paraganglioma/pheochromocytoma syndrome. The stenosing rectosigmoid neoplasm could also have been an intestinal stromal tumor (GIST), which is associated with such a syndrome, although tumor location was more consistent with an intestinal adenocarcinoma. Based on this, measurements were made of fractionated catecholamines and metanephrines in 24-h urine (epinephrine <10μg/24h [0–25], dopamine 204μg/24h [0–400], normetanephrine >1171μg/24h [0–500], norepinephrine 733μg/24h [0–80], metanephrine 55μg/24h [0–300], calcitonin <2pg/m [0–18.2], and chromogranin A 1904.2ng/mL [19.4–48.1]). [131I]Metaiodobenzylguanidine scintigraphy showed a highly increased uptake in lesions of both the adrenal glands and the retroperitoneal mass (Fig. 1), while tumor extension scintigraphy with [111In]octreotide showed an increased uptake at the right tympanic, right carotid, mediastinal, and lumbar paravertebral levels (Fig. 2), and a genetic study for the SDH mutation revealed the c.197G≥A/p.W66X mutation in heterozygosis in exon 2 of gene 11q23 of SDH subunit D, consisting of the substitution of adenine for guanine in nucleotide 197 of cDNA. This mutation has previously been reported and has been shown to be associated with the disease.4


After α-adrenergic blockade with phenoxybenzamine, surgical resection was performed of both adrenal glands, the retroperitoneal mass, and the rectosigmoid junction tumor.
A histological study found two 9-mm and 19-mm pheochromocytomas, a 20-mm extra-adrenal paraganglioma, and a low-grade adenocarcinoma infiltrating perirectal fat, lymphatic vessels, and nerve tracts (T3N2M0).
Two months after surgery, the patient showed normal blood glucose without treatment, high blood pressure, normal urinary metanephrine and chromogranin A levels, and persistently high CEA levels. Nutritional support through percutaneous endoscopic gastrostomy was required. An abdominal CT scan showed two hepatic lesions suggesting metastasis, while [111In]octreotide scintigraphy revealed no hepatic or abdominal uptake. However, prior mediastinal and jugulotympanic uptake persisted, which, together with the markers, suggested the progression of colonic adenocarcinoma. The patient was therefore referred for oncological follow-up.
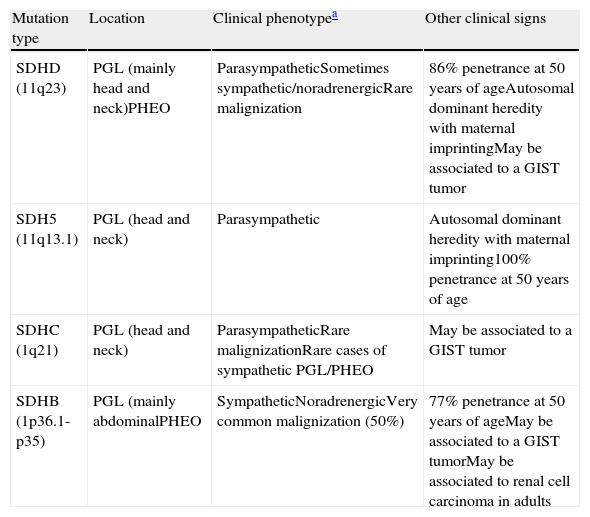
SDH-related paraganglioma/pheochromocytoma syndromes are inherited as an autosomal dominant condition with incomplete penetrance and cause multiple paragangliomas in different locations. SDH is part of complex II of the mitochondrial respiratory chain. SDH consists of four subunits encoding for four nuclear genes, SDHA, SDHB, SDHC, and SDHD. Different mutations in each of these subunits will result in different genotypes-phenotypes and are responsible for 80% of familial clusters of PGL and PHEO5 (Table 1). The other hereditary diseases where pheochromocytoma and paraganglioma may be associated include MEN 2, von Hippel-Lindau, and neurofibromatosis type 1. These were ruled out in our patient because they have other associated characteristics which may be recognized by clinical examination and supplemental tests and which were not found in the patient.
Characteristics of SDH mutations.
| Mutation type | Location | Clinical phenotypea | Other clinical signs |
| SDHD (11q23) | PGL (mainly head and neck)PHEO | ParasympatheticSometimes sympathetic/noradrenergicRare malignization | 86% penetrance at 50 years of ageAutosomal dominant heredity with maternal imprintingMay be associated to a GIST tumor |
| SDH5 (11q13.1) | PGL (head and neck) | Parasympathetic | Autosomal dominant heredity with maternal imprinting100% penetrance at 50 years of age |
| SDHC (1q21) | PGL (head and neck) | ParasympatheticRare malignizationRare cases of sympathetic PGL/PHEO | May be associated to a GIST tumor |
| SDHB (1p36.1-p35) | PGL (mainly abdominalPHEO | SympatheticNoradrenergicVery common malignization (50%) | 77% penetrance at 50 years of ageMay be associated to a GIST tumorMay be associated to renal cell carcinoma in adults |
Sympathetic tumors are functioning and secrete catecholamines. Parasympathetic tumors are non-functioning. Head and neck paragangliomas are usually parasympathetic, while pheochromocytomas and abdominal paragangliomas are sympathetic. Almost all noradrenergic tumors secrete norepinephrine and normetanephrine only, while adrenergic tumors secrete epinephrine and metanephrine in addition to norepinephrine and normetanephrine.
The course of the disease is relatively benign. It is only transmitted by the father, with maternal “imprinting”: when the mutation is inherited from the mother. Her children carry the mutation but do not develop the disease.
Metanephrine measurement is the best biochemical test for diagnosing functioning pheochromocytoma or paraganglioma. Free or total plasma metanephrines and fractionated metanephrines in 24-h urine have diagnostic sensitivities (SE) of 96%, 95%, and 95%, respectively and specificities (SP) of 89%, 91%, and 86%, respectively.6
CT and MRI (having 98% SE and 70% SP) are the first-choice location tests, although MRI appears to provide a greater diagnostic yield in extra-adrenal tumors.7
[131I]Metaiodobenzylguanidine (MIBG) scintigraphy continues to be the first choice test for the location of secretory pheochromocytoma because it has a high SE (94%) and SP (92%) and allows for the confirmation of the secretory nature of the tumor, for the location of tumors not seen in CT/MRI, and for the identification of other disease sites.8
Tumor extension [111In]octreotide scintigraphy shows in the series SE values of 88–97% and SP values of 75–82%, particularly in patients with suspected metastatic or extra-adrenal disease.
New radiotracers have recently been developed, including fluorodopamine ([18F]-DOPA), fluorodihydroxyphenylalanine ([18F]-FDA), fluorodeoxyglucose ([18F]FDG), or dota-Nal octreotide (DOTATOC). [18F]-DOPA PET appears to have a greater diagnostic value for the detection of non-metastatic extra-adrenal PGLs as compared to [111In]octreotide, with a 95.7% sensitivity, and has a greater diagnostic yield, mainly in PGLs ≤10mm and in abdominal PGLs. PGLs of the head and neck are more easily recognized in both tests.9
As occurred in our case, several complementary location tests should be used to optimize the diagnosis of these syndromes.
Please, cite this article as: Sánchez-Pacheco Tardón M, et al. Diagnóstico tardío de un caso índice de síndrome paraganglioma/feocromocitoma asociado a la SDH. Endocrinol Nutr. 2012;59:148–50.
