


Adrenal myelolipoma is a benign, rarely functioning tumor consisting of adipose tissue and myeloid cells in different proportions. Adrenal myelolipoma accounts for approximately 6% of adrenal incidentalomas1 and is found in up to 0.2% of autopsy studies.2 Few cases of myelolipomatous masses causing Cushing's syndrome3 (CS) have been reported in the literature, so that the functioning capacity that these tumors may sometimes have can easily be overlooked. The case of a patient with clinically evident CS in whom imaging tests disclosed an adrenal myelolipoma is reported below.
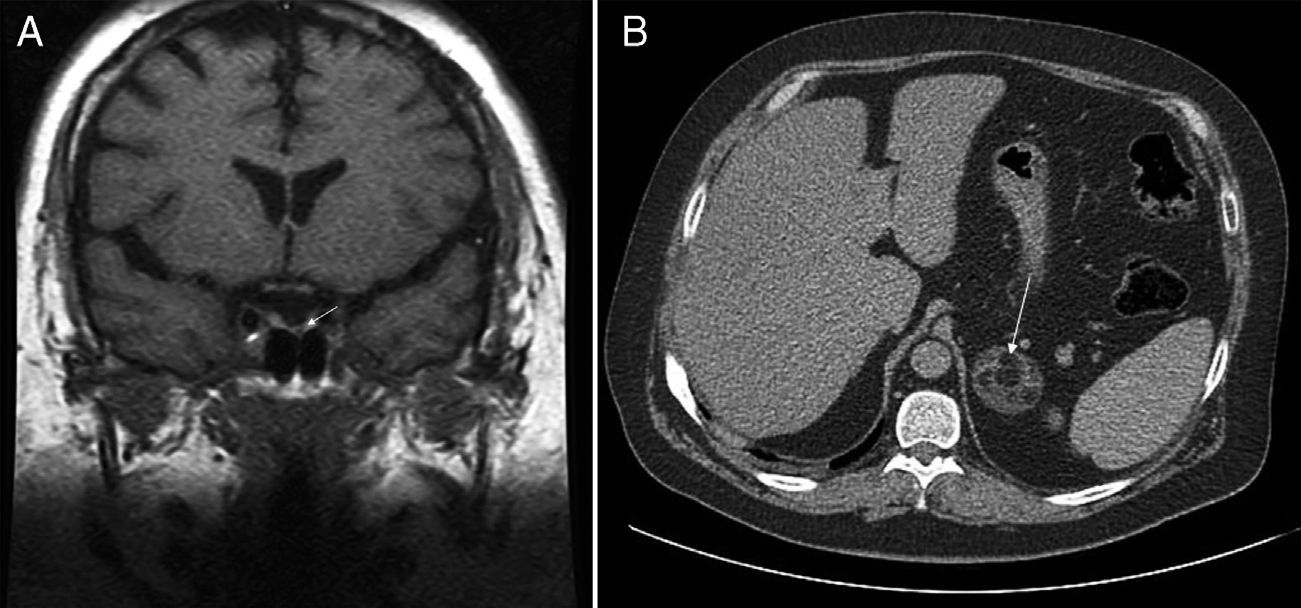
This was a 36-year-old male with diabetes who attended our hospital for blood glucose monitoring. The patient had been diagnosed 10 years before with metabolic syndrome with high blood pressure, type 2 diabetes, dyslipidemia, and obesity. The course of the disease had been unfavorable despite compliance with hygienic and dietary advice and with adequate medication. Physical examination revealed central obesity with a body mass index (BMI) of 38.6kg/m2, waist circumference of 125cm, blood pressure of arterial 149/109mmHg, and clinical features of frank CS with facial redness, red-wine abdominal striae, and strong muscle weakness. Basal plasma ACTH levels were suppressed (4.5pg/mL; normal range [NR] 5–45), while 24-h plasma cortisol levels (15mcg/dL) and 24-h urinary free cortisol levels (1218mcg; NR 4.3–176) were both elevated. Plasma cortisol was not suppressed (16mcg/dL) after administration of dexamethasone 8mg. Abdominal computed tomography (CT) showed a left adrenal mass 4.5cm in the longest axis with a fat density component suggesting myelolipoma (Fig. 1A). Pituitary magnetic resonance imaging (MRI) disclosed a 2-mm lesion in the left medial region of the pituitary gland (Fig. 1B). Suppressed plasma ACTH levels and lack of cortisol suppression after high dexamethasone doses ruled out the pituitary nodule as the cause of CS and identified it as an incidentaloma of the same origin. Surgery was performed to resect the myelolipoma in the left adrenal gland. The postoperative plasma cortisol level was <0.1mcg/dL, and corticoid replacement therapy was therefore required.
 Pituitary MRI: left paramedial hypointense nodular image 2mm in size (arrow) in the sequence without contrast acquired in the coronal plane. (B) Abdominal CT: left adrenal nodule 4.8cm in longest axis, with fat density component, suggesting myelolipoma (arrow).")
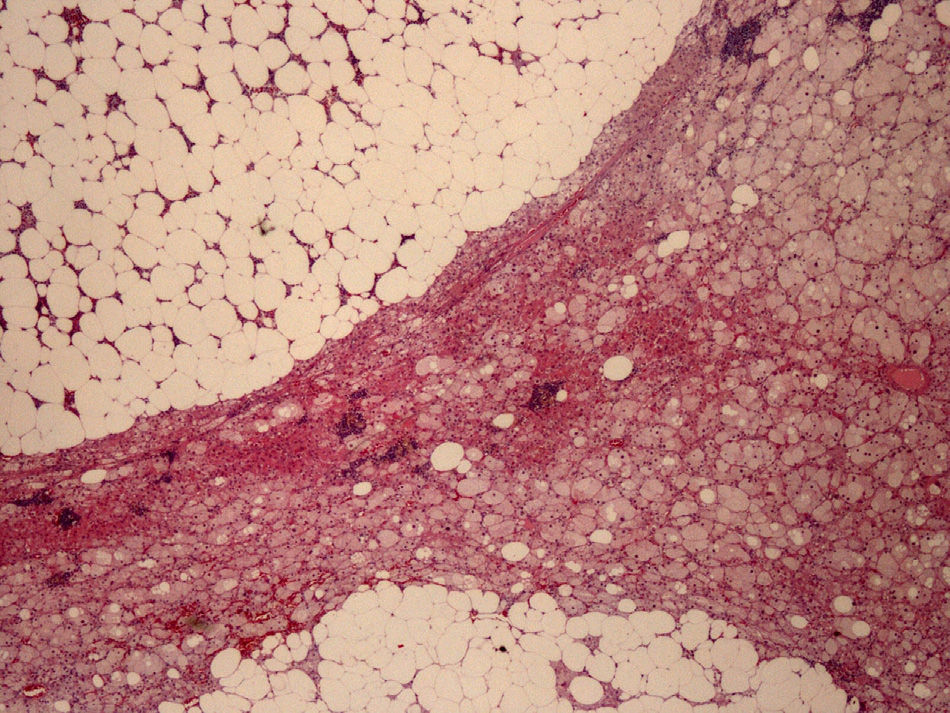
The left adrenalectomy specimen consisted of a heterogeneous orange ovoid formation, 4.5cm×4cm in size, with abundant yellowish brown areas. The normal adrenal gland was compressed and peripherally reduced, with a largest diameter of 0.4cm. Histologically, the neogrowth had well circumscribed margins and consisted of two components (Fig. 2): the adrenocortical adenoma or adenomatous component (30% of the tumor) and the myelolipomatous component (70% of the tumor). The first component formed cords of oxyphilic clear cells, with sometimes vacuolated cytoplasm, due to their lipid contents and nuclei with marked focal atypia but low mitotic activity, with no necrosis, myxoid changes or vascular invasion. The second component consisted of mature adipocytes and cell elements from all three hemopoietic series in different maturation phases.
.")
Immunohistochemistry, used to confirm diagnosis and for differentiation from pheochromocytoma and adrenal or renal carcinoma, amongst other potential tumors, was positive for inhibin and melan-A (adrenocortical markers), focal with synaptophysin, but negative for chromogranin A (adrenomedullary marker), cytokeratin 7, and CD10 (positive markers in metastatic carcinomas of renal origin). Ki 67 was expressed in the nucleus of 1–2% of the tumor cells as a sign of their low proliferative activity. The pathological diagnosis was adrenocortical adenoma with extensive myelolipomatous metaplasia.
Adrenal myelolipoma is considered a benign and non-secreting tumor. It may however cause severe CS lasting several years and easily resolved after adrenalectomy. This association has been reported in only 10 cases with different clinical characteristics from the one reported here, as they were mild CS with little symptoms, more common in women and with a short course. Adrenalectomy resolved the disease in all cases. Our patient also had a pituitary incidentaloma. This is a finding made in approximately 10% of the general population when MRI of the brain is performed for another reason.4
The pathogenesis of these myelolipomas is highly controversial. Most hypotheses are based on the idea that, rather than being true neoplasms, these are of metaplastic origin, resulting from different types of stimuli, including the hyperproduction of cortisol, aldosterone, or androgens by the adrenocortical neoplasm or hyperplasia.5 Myelolipoma has also been reported as being associated with hormone hyperproduction syndromes such as pheochromocytoma, Conn's syndrome, and congenital adrenal hyperplasia due to 21-hydroxylase deficiency.6–8 Histologically, it results from the combination of mixed elements of adrenal cells with secretory capacity together with myeloid cells and fat metaplasia and, as occurred in our patient, adenoma cells (but not adipocytes or hemopoietic precursors) are responsible for hormone synthesis. On the other hand, cell culture studies suggest a novel mechanism of immunoendocrine interaction in which the direct contact of adrenal cortex cells with purified T lymphocytes mediates the stimulation of the secretion of hormones such as dehydroepiandrosterone (DHEA and cortisol).9
Clinical practice guidelines on the management of adrenal incidentaloma10 do not recommend hormone assessment in pure myelolipomas, but differentiating these from a mixed lesion is not always easy. We therefore think that hormone assessment is needed in any adrenal mass because even those with a radiographic appearance of myelolipoma may be secretory, particularly when they are heterogeneous in CT or MRI and the clinical signs so suggest.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Corpas Jiménez MS, Ortega Salas R, Tenorio Jiménez C, Molina Puerta MJ. Mielolipoma asociado a adenoma adrenocortical: una causa infrecuente de síndrome de Cushing. Endocrinol Nutr. 2014;61:e7–e9.
