





Non-alcoholic fatty liver disease (NAFLD) is an aberrant lipid metabolism disease. Hypoxia inducible factor-1 (HIF-1α) is a transcription factor which plays an important part in adapting lower oxygen condition. Here, we aimed to clarify the relationship between HIF-1α and NAFLD.
MethodsHepG2 cells was stimulated by oleic acid (OA) and palmitic acid (PA) to establish in vitro model of NAFLD. The expression of lipid metabolism-related genes, the binding of PPARα to HIF-1α promoter, the lipid deposition, and oxidative stress were detected by qRT-PCR, western blot, Chip assay, Oil Red O staining and ELISA assays, respectively.
ResultsHIF-1α silence promoted lipid accumulation in NAFLD cells, accompanying by the significantly increased contents of TG (triglyceride) and ApoB (apolipoprotein B). In HepG2 cells treated with OA/PA, the expression of lipid metabolism-related genes and proteins, including APOE, A2m, TNFRSF11B, LDLr, and SREBP2, and the intracellular lipid deposition were up-regulated and further aggravated after silencing HIF-1α. In addition, the loss of HIF-1α could remarkably elevate MDA contents while inhibit the activities of beneficial antioxidant enzymes SOD and GSH-Px to activate oxidative stress, and promote the secretion of pro-inflammatory IL-6 and TNF-α to aggravate inflammation in NDFLD cells. PPARα positively bound to HIF-1α promoter. The silence of PPARα aggravated lipid deposition under normal or hypoxic environment in NAFLD cells. In addition, PPAR-α silence could decrease the expression of HIF-1α and ANGPTL4 in NAFLD cell model; moreover, the expression of APOE, A2m and TNFRSF11B and the production of TG and MDA were increased by PPAR-α suppression.
ConclusionHIF-1α plays a crucial role in the regulation of lipid metabolism through activating PPAR-α/ANGPTL4 signaling pathway in NAFLD.
La esteatohepatitis no alcohólica (EHNA) es una enfermedad del metabolismo aberrante de los lípidos. El factor inducible por hipoxia 1 (HIF-1α) es un factor de transcripción que desempeña una función importante en la adaptación de la afección de nivel de oxígeno bajo. En el presente documento, intentamos aclarar la relación entre HIF-1α y la EHNA.
MétodosLas células HepG2 se estimularon con ácido oleico (OA) y ácido palmítico (PA) para establecer un modelo in vitro de la EHNA. La expresión de los genes relacionados con el metabolismo de los lípidos, la unión de PPARα al promotor HIF-1α, el depósito de lípidos y el estrés oxidativo se detectaron mediante ensayos de qRT-PCR, inmunoelectrotransferencia, ensayo de inmunoprecipitación de cromatina (ChIP), ensayos de tinción de rojo aceite O y ELISA, respectivamente.
ResultadosEl silencio de HIF-1α promovió la acumulación de lípidos en las células de la EHNA, acompañada de un aumento significativo del contenido de triglicéridos (TG) y apolipoproteína B (ApoB). En las células HepG2 tratadas con OA/PA, la expresión de genes y proteínas relacionados con el metabolismo lipídico, incluidos APOE, A2m, TNFRSF11B, LDLr y SREBP2, y el depósito de lípidos intracelular se regularon al alza y se agravaron aún más después de silenciar HIF-1α. Además, la pérdida de HIF-1α podría elevar notablemente el contenido de MDA e inhibir las actividades de las enzimas antioxidantes beneficiosas SOD y GSH-Px para activar el estrés oxidativo, y promover la secreción de IL-6 pro-inflamatoria y TNF-α para agravar la inflamación en las células de la EHNA. PPARα se unió positivamente al promotor HIF-1α. El silencio de PPARα agravó el depósito de lípidos en un ambiente normal o hipóxico en las células de la EHNA. Además, el silencio de PPAR-α pudo disminuir la expresión de HIF-1α y ANGPTL4 en el modelo de células de la EHNA; por otra parte, la expresión de APOE, A2m y TNFRSF11B, y la producción de TG y MDA aumentaron por la supresión de PPAR-α.
ConclusiónHIF-1α desempeña una función crucial en la regulación del metabolismo de los lípidos a través de la activación de la vía de señalización PPAR-α/ANGPTL4 en la EHNA.
Non-alcoholic fatty liver disease (NAFLD) is an epidemic chronic liver disease that characterized by abnormal lipid deposition in hepatocytes in the absence of excess alcohol intake.1,2 NAFLD include a wide spectrum of liver pathologies, which range from simple steatosis through steatohepatitis to cirrhosis.3 It steadily increases along with the worldwide epidemic of obesity and type 2 diabetes.4,5 NAFLD is a multisystem disease and its clinical burden is not only involves several extrahepatic dysfunctions but also limits liver-related morbidity and mortality.6–8 Some research show that the mark of NAFLD is a triglycerides-rich lipid droplets over-accumulation in hepatocytes, which due to hepatic lipid metabolism deregulation.9 Triglycerides and lipotoxic lipids accumulate in liver and lead to serious liver injuries through activating oxidative stress, inducing cell death and inflammatory responses.8,10 In order to prevent and therapy NAFLD, some cellular and molecular mechanisms are investigated.
PPARα, a ligand-activated nuclear receptor, is the molecular target of inducing peroxisome proliferation and highly express in the liver.11 Although it is also expressed in many tissue and cells including intestine, smooth muscle and immune cells, it mainly express in tissue that enrich high fatty acid oxidation rates such as liver.12 PPARα can response to feeding and starvation through regulating the rate of fatty acid catabolism and lipogenesis and ketone body ratio.13 PPARα play an important part in regulating peroxisome and mitochondrial β-oxidation, fatty acid transport and hepatic glucose production.14 Some research illustrate that PPARα can negatively regulate pro-inflammatory and acute phase response signaling pathway in systemic inflammation, atherosclerosis and non-alcoholic steatohepatitis models.15 Angiopoietin-like (ANGPTL4) protein is a multifunction secreted protein and regulates LPL activity by directly interacting with LPL and preventing LPL dimerization.16 Moreover, It regulates lipid metabolism, glucose homeostasis and atherosclerosis progression.17 Some reports show that targeting PPARβ/δ to inhibit ANGPTL4 expression may be an alternative approach for treating metabolic disorder-associated HNSCC.18
Several studies have showed that liver injury can cause tissue hypoxia through changing the liver perfusion or mitochondrial modification.19 Hypoxia-inducible factors (HIFs) play an important role in mediating homeostatic response to hypoxia.20 HIF is a transcription factor consisted of two basic helix-loop-helix proteins of the PAS family – a hypoxia regulated subunit (HIF-1α or HIF-2α) and a constitutive β-subunit (HIF-1β).21,22 Some studies have reported that HIF is closely related with NAFLD.19 Mesarwi et al. have shown that HIF-1α is a key factor in hypoxia promoting the development of liver fibrosis in NAFLD.23 Some publications have demonstrated that tissue hypoxia can increase fibro genic enzyme Lysol oxidase (LOX) expression through regulating HIF-1.24 Additionally, some studies have showed that HIFs suppress PPARα-EGF21-Nrf2 endocrine signaling pathway against oxidative stress and acetaminophen-induced toxicity in liver.25,26 Although between HIF and hepatic PPARα activity have certain implication in liver toxicity, whether HIF has closely relationship with PPARα in NAFLD still explored.
In the current study, we utilized HepG2 cells stimulated with OA/PA to establish NAFLD cell model to clarify the relationship between HIF-1α and PPAR-α/ANGPTL4 signaling pathway during NAFLD. Our data demonstrated that HIF-1α silencing could inhibit PPAR-α and ANGPTL4 expression and aggravate lipid metabolism in NAFLD. It suggests that HIF-1α can become a new target in NAFLD treatment.
Materials and methodsCells culture and treatmentThe human hepatocellular carcinoma line HepG2 was purchased from ATCC (wManassas, VA, USA) and cultured in RPMI-1640 medium (Gibco, Vienna, NY, USA) or DMEM high glucose medium (Gibco, Vienna, NY, USA) supplemented with 10% FBS, 100U/mL penicillin, and 100μg/ml streptomycin. HepG2 cells transfected with pLKO.1 lentiviruses expressing shRNAs targeting mRNAs of HIF1α (sh-HIF-1α) or PPARα (sh-PPARα) were treated with or without 400μM OA and PA for 48h prior to follow-up studies. shRNA or the corresponding sh-Scramble (sh-NC) were purchased from Ribobio (Guangzhou, China).
Quantitative real-time PCR analysisCells from different treatment groups were collected and subjected to total RNA extraction using RNAiso Plus (Takara, 9108). RNA samples were reverse transcripted into cDNA by PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, RR047A). Quantitative real-time PCR was then performed on a MyiQ single-color RT-PCR detection system by using SYBR® Premix Ex Taq (Takara, RR820A) with the amplification conditions listed as follows: 95°C, 10min for denaturation; 95°C, 5s and then 60°C, 1min for 40 cycles; 95°C, 15s, 60°C, 1min, and 95°C, 15s, for melt curve. All primer sequences in this study are listed in Table 1. The relative mRNA expression was normalized to that of GAPDH and determined using the comparative Cq method (2−ΔΔCq).
Primer sequences used in this study.
| Primer name | Sequence (5′ to 3′) |
|---|---|
| HIF-1α | (F) 5′-CCC ATT CCT CAC CCA TCA AAT A-3′ |
| (R) 5′-CTT CTG GCT CAT ATC CCA TCA A-3′ | |
| VLDL | (F) 5′-CAG GAA GAT TGG CTT AGA GAG G-3′ |
| (R) 5′-GCT TAG ATC GGC CCA GAA TAG-3′ | |
| CPT-1 | (F) 5′-TGG GAA ACA AGC CAG AAG AA-3′ |
| (R) 5′-GAA GCT CCC ACT GCC ATA AA-3′ | |
| APOE | (F) 5′-GAA GGC CTA CAA ATC GGA ACT-3′ |
| (R) 5′-GCA CAC GTC CTC CAT GTC-3′ | |
| A2m | (F) 5′-TGA AGA TGC TGG ACT TGG TAT C-3′ |
| (R) 5′-CTC CAC GAA TCA CAG AGT AAG G-3′ | |
| TNFRSF11B | (F) 5′-GTC TTT GGT CTC CTG CTA ACT C-3′ |
| (R) 5′-CCT CAC ACA GGG TAA CAT CTA TTC-3′ | |
| PPAR-α | (F) 5′-CCT GCA AGA AAT GGG AAA CAT C-3′ |
| (R) 5′-GCC AGG ACA GCT TCC TAA AT-3′ | |
| ANGPTL4 | (F) 5′-GAG GCT GGA CAG TAA TTC AGA G-3′ |
| (R) 5′-CGT GAT GCT ATG CAC CTT CT-3′ | |
| LDLr | (F) 5′-AAT GCT TGG ACA ACA ACG GC-3′ |
| (R) 5′-CGG GAT CCT GAC ACT CAT CG-3′ | |
| mTORC1 | (F) 5′-CGA TGG CCA GGG ATC TCT TC-3′ |
| (R) 5′-GGT CTG TGT GAC TTC AGC GA-3′ | |
| SREBP2 | (F) 5′-ATC GAC CTA GGC AGT CTG GT-3′ |
| (R) 5′-ATA GAG GGC TTC CTG GCT CA-3′ | |
| GAPDH | (F) 5′-GTG GTC TCC TCT GAC TTC AAC-3′ |
| (R) 5′-CCT GTT GCT GTA GCC AAA TTC-3′ |
For western blot analysis, HepG2 cells from different treatment groups were lysed in RIPA buffer containing protease inhibitor cocktail (Generay, Shanghai, China). After quantifying protein concentration using a BCA Assay Kit (Thermo Scientific), protein samples were loaded and separated by 10% SDS-polyacrylamide gels, electro-transferred to polyvinylidene fluoride membranes, blocked with 5% BSA and then incubated with the specific primary antibodies, including anti-HIF-1α, anti-A2m, anti-APOE, anti-TNFRSF11B, anti-PPARα, anti-ANGPTL4, anti-LDLR, anti-SREBP2, anti-phospho-mTOR, anti-mTOR, and anti-GAPDH (Cambridge, MA, USA). After washing with TBST, the blots were treated with HRP-conjugated secondary antibody and finally detected using Bio-Imaging System and Quality One 1-D analysis software (Bio-Rad, Richmond, CA, USA).
Oil Red O stainingHepG2 cells transfected with sh-HIF-1α or sh-PPARα were stimulated with or without OA/PA and then stained with Oil Red O (5mg/ml; St. Louis, USA) for 20min at 37°C to visualize the intracellular lipid droplets. Images were acquired with Inverted fluorescence Microscope (Shinagawa-ku, Tokyo, Japan laser).
Measurements of triglyceride (TG) level and oxidative stress biomarkersHepG2 cells with different treatment were harvested and suspended with PBS. Then the cell suspension was homogenized and broken by ultrasound in ice to measure TG level by using triglyceride assay kit (Nanjingjiancheng, Nanjing, China), and to detect oxidative stress biomarkers including maleic dialdehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) with malondialdehyde assay kit, superoxide dismutase activity assay kit and glutathione peroxidase assay kit purchased from Nanjing Jiancheng Bioengineering Institute (China). The protein concentration was measured using a BCA assay kit (Beyotime Biotechnology, Shanghai, China).
ELISA detection for the secretion of cytokines and apolipoproteinsTo detect the secretion of cytokines and apolipoproteins, the cell supernatant from different treatment groups were collected. The contents of ApoE and ApoB in the culture medium of HepG2 were measured using Human ApoE ELISA kit (Cusabio, CSB-EL001936RH) and Human ApoB ELISA kit (Cusabio, CSB-E08100h), respectively, according to the manufacturers protocol. In addition, the secretion levels of IL-1β and TNF-α were also determined by Human IL-1β and Human TNF-α ELISA kits (cusabio, CSB-E08053h# and CSB-E04740h#).
ChiP assayChiP was performed by using the Sample ChiP Enzymatic Chromatin IP kit (Millipore, Massachusetts, America) according to the manufacturer's instructions. HepG2 cells were isolated by the kit (Millipore). Immunoprecipitation was performed with the antibody against PPARα, IgG and Histone H3. IgG was used as a control and Histone H3 as the positive control. Precipitated DNAs were identified by PCR using specific primers that detects the binding of PPARα to promoter of HIF-1α gene: HIF-1α, forward: 5′-CCC ATT CCT CAC CCA TCA AAT A-3′, reverse: 5′-CTT CTG GCT CAT ATC CCA TCA A-3′.
Statistical analysisAll data were statistical analysised by Student's t-test using Graph pad prism 4.0 (Graph pad Software, La Jolla, CA). P<0.05 was considered statistically significant.
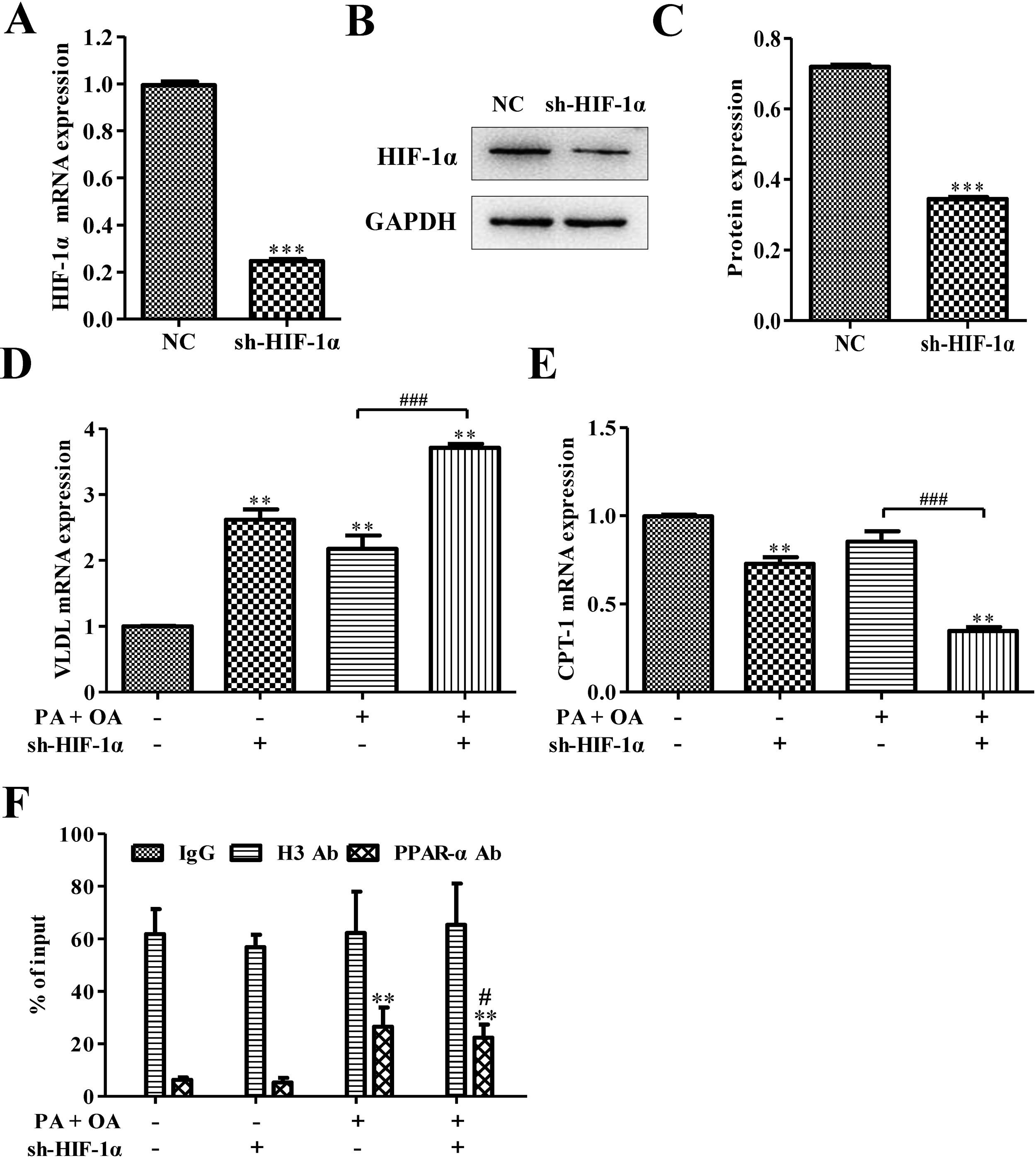
ResultsSilencing HIF-1α inhibited PPARα signaling pathwaySome reports show that HIFs correlate inversely with the expression of PPARα in hepatocytes.21 In order to investigate how PPARα affects HIF-1α, sh-HIF-1α was used to silence HIF-1α in HepG2 cells. The results showed that the expression levels of HIF-1α mRNA and protein were all significantly down-regulated after silencing HIF-1α (Fig. 1A–C). The mRNA level of very low-density lipoprotein (VLDL) receptor, which accelerated hepatic TG overload in HepG2 cells and enhanced apoprotein B secretary protein supporting the excretion TG from hepatic cells in experimental models induced by palmitate and high-fat diet,27 was increased under OA/PA stimulation and furtherly up-regulated by sh-HIF-1α treatment (Fig. 1D). Once activated, PPARα induces fatty acid oxidation-related genes, such as CPT-1.28 Here, in hepatocytes exposed to OA and PA, CPT-1 mRNA expression had been inhibited, suggesting a reduction of beta-oxidation of fatty acids which is closely associated with the transcriptional control of genes by PPARα,28 see Fig. 1E. Interestingly, HIF-1α silence could further repress CPT-1 mRNA expression, indicating that there may be some relationship between HIF-1α and PPARα on regulating NAFLD. Results from Chip assay showed that, compared with normal cells, the physical binding of PPARα to the HIF-1α gene promoter was enriched in NAFLD cells. Furthermore, the binding of PPARα with HIF-1α promoter was declined after HIF-1α was silenced (Fig. 1F). These findings revealed that HIF-1α had a positive correlation with PPARα in both normal hepatocytes and NAFLD cells, and the silence of HIF-1α could decrease CPT-1 but increase VLDL mRNA expression. It suggests that HIF-1α may affect NAFLD via PPARα signaling pathway.
 The mRNA expression level of HIF-1α was down-regulated after silencing HIF-1α; Consistent with this finding, silencing HIF-1α could decrease the protein expression of HIF-1α (B). The protein was quantified using densitometry (C). In NAFLD cells, qRT-PCR assay for the mRNA expression of VLDL (D) and CPT-1 (E), which is regulated by PPAR-α signaling pathway. (F) The physical binding of PPAR-α to the HIF-1α gene promoter was enriched in NAFLD cells as shown by the result of ChiP assay. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.")
Silencing HIF-1α inhibited PPAR-α signaling pathway. (A) The mRNA expression level of HIF-1α was down-regulated after silencing HIF-1α; Consistent with this finding, silencing HIF-1α could decrease the protein expression of HIF-1α (B). The protein was quantified using densitometry (C). In NAFLD cells, qRT-PCR assay for the mRNA expression of VLDL (D) and CPT-1 (E), which is regulated by PPAR-α signaling pathway. (F) The physical binding of PPAR-α to the HIF-1α gene promoter was enriched in NAFLD cells as shown by the result of ChiP assay. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.
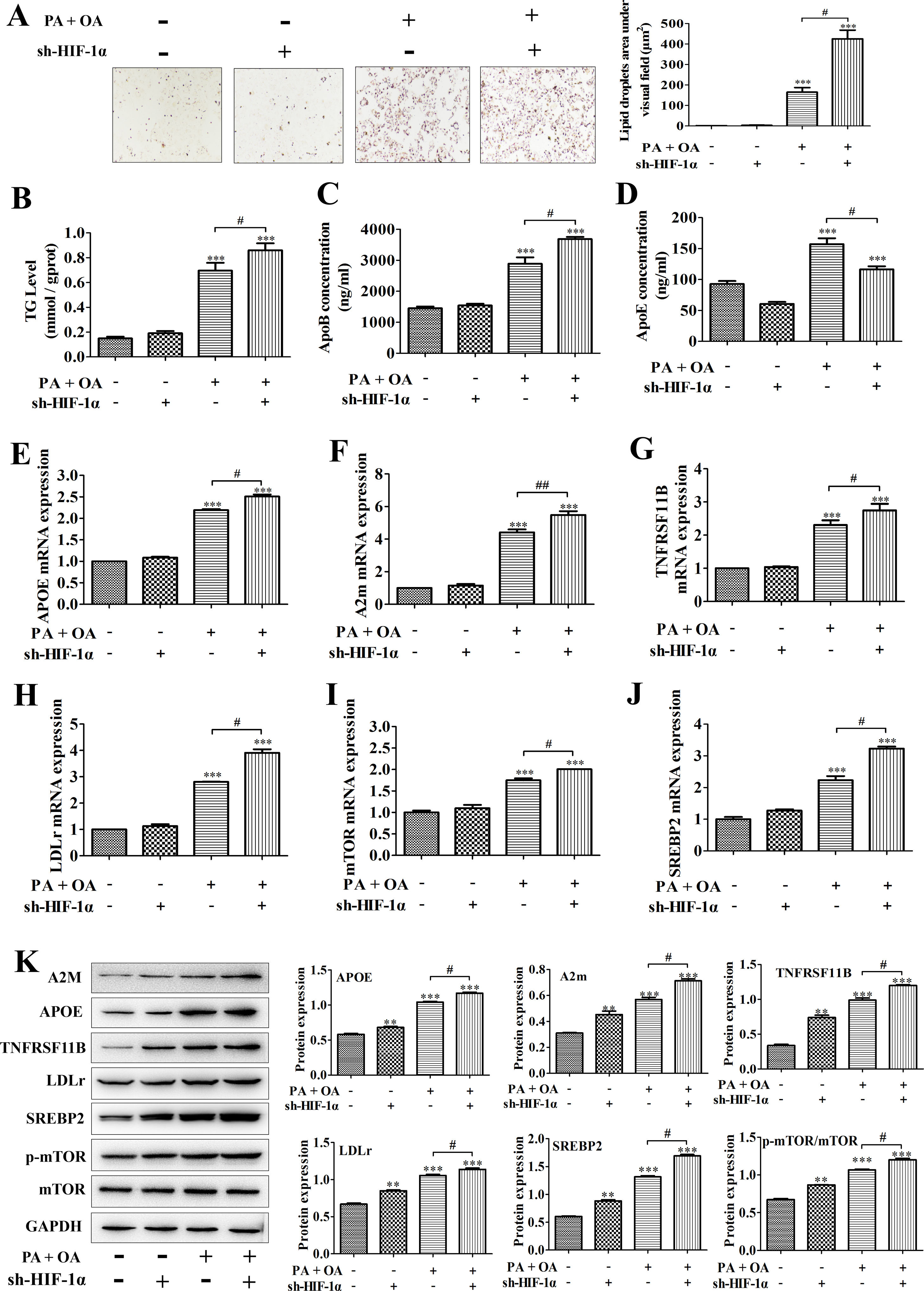
A model of hepatic steatosis was established in cultured hepatocytes to explore the role of HIF-1α in NAFLD in vitro. Compared with normal hepatocytes, intracellular lipid accumulation was increased in OA/PA stimulated hepatocytes, reflecting as the results from Oil Red O staining; after silencing HIF-1α, the number of lipid droplets in OA/PA treated hepatocytes were markedly increased (Fig. 2A). Moreover, consistent with this finding, the contents of TG and ApoB, which are positively correlated with NAFLD incidence, in the culture medium of OA/PA-treated HepG2 cells were also elevated significantly with sh-HIF-1α transfection (Fig. 2B, C). However, ApoE concentration was decreased by sh-HIF-1α silence both in normal and NAFLD cells, especially in NAFLD cells (Fig. 2D). To explore the regulatory effect of HIF-1α on the expression of lipid metabolism-related genes during NAFLD, we performed qRT-PCR and found that, under OA/PA stimulation, the mRNA expression levels of APOE, A2m, LDLr, mTOR, SREBP2 and TNFRSF11B were all increased when compared with those of normal cells; however, HIF-1α silence could further un-regulate the expression of these genes in NAFLD cells, but it had no significant effect on the expression of these lipid metabolism-related genes in normal HepG2 cells (without OA/PA stimulus), see Fig. 2E–J. Furthermore, the result from western blot just displayed a consistent trend toward that of qRT-PCR assay, that is, OA/PA stimulus elevated the protein expression levels of lipid metabolism-related genes, such as LDLr, SREBP-2, ApoE and A2m, which are closely associated with the fatty-acid transport and the lipogenesis. This effect was more prominent in NAFLD cells (Fig. 2K). The activation of mTOR signaling pathway can up-regulate the mRNA expression of HIF-1α and thus elevated the production of HIF-1α.29 In our study, the phosphorylation level of mTOR was increased by HIF1-α silence, indicating that the deletion of HIF-1α in HepG2 cells can activate mTOR pathway competitively by up-regulating HIF-1α mRNA expression and increasing mTOR phosphorylation (Fig. 2K). Taken together, these data showed that HIF-1α silence may contribute to hepatic steatosis by regulating lipid metabolism-related genes, suggesting that activating HIF-1α could turn into a treatment strategy for NAFLD in clinic.
 the lipid accumulation was detected by Oil Red O staining; right, the lipid droplets area under visual field; (B–D) the contents of TG, ApoB and ApoE in cell homogenate were determined by triglyceride assay kit or ELISA kits, respectively; (E–J) the mRNA expression levels of APOE, A2m, TNFRSF11B, LDLr, mTOR, and SREBP2 were respectively quantified using qRT-PCR; (F) the protein expression levels of these lipid metabolism-related genes were measured by western blotting; meanwhile, the proteins were quantified using densitometry. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 vs NAFLD cells without HIF-1α silence.")
Silencing HIF-1α increased lipid deposition in NAFLD cells by regulating the expression of lipid metabolism-related genes. In NAFLD cells treated with or without sh-HIF-1α, (A) the lipid accumulation was detected by Oil Red O staining; right, the lipid droplets area under visual field; (B–D) the contents of TG, ApoB and ApoE in cell homogenate were determined by triglyceride assay kit or ELISA kits, respectively; (E–J) the mRNA expression levels of APOE, A2m, TNFRSF11B, LDLr, mTOR, and SREBP2 were respectively quantified using qRT-PCR; (F) the protein expression levels of these lipid metabolism-related genes were measured by western blotting; meanwhile, the proteins were quantified using densitometry. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 vs NAFLD cells without HIF-1α silence.
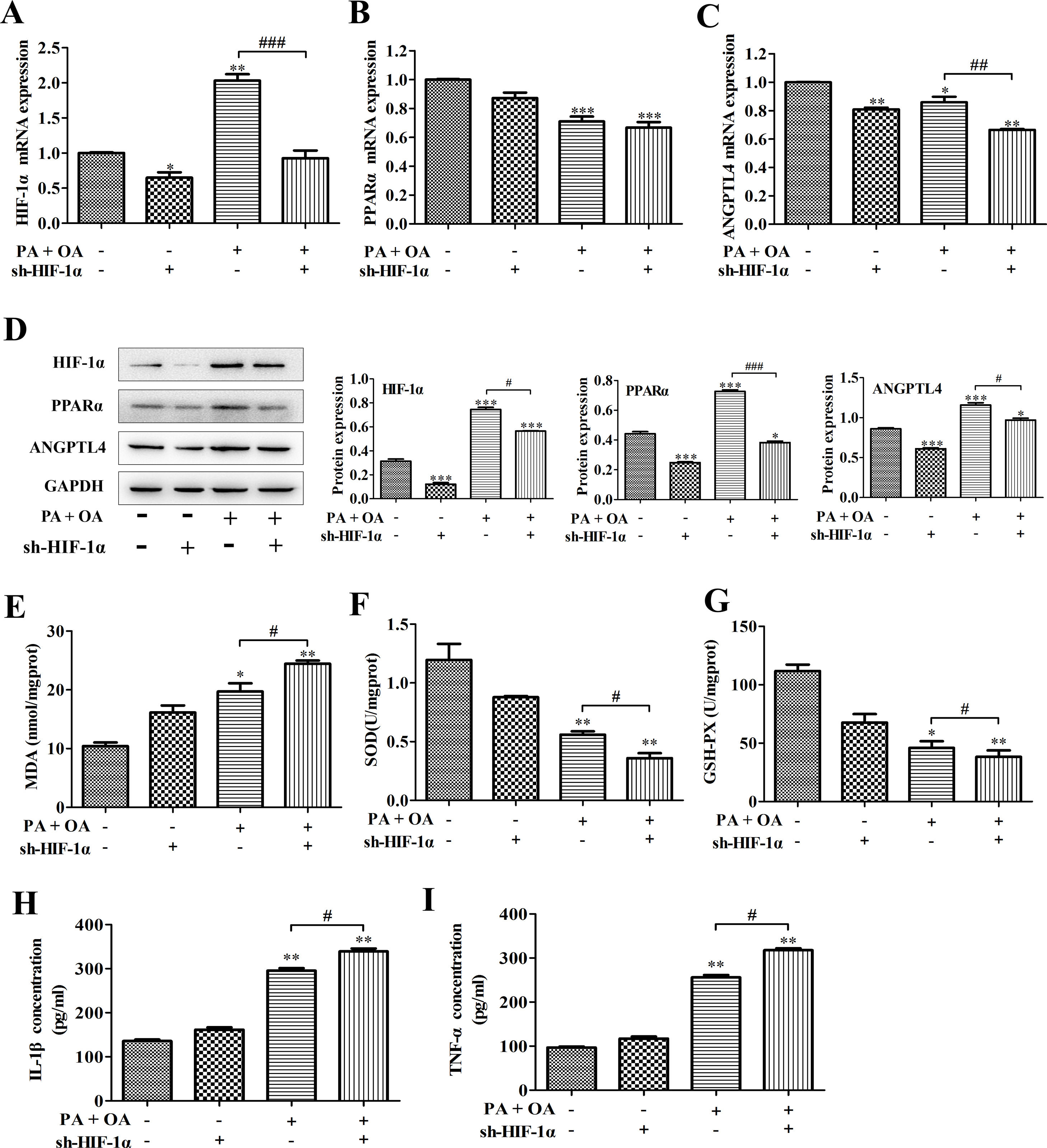
It has been reported that unchanged or even down-regulated PPARα were observed in NAFLD.30 Similarly, in this study, a significant decrease of PPARα and ANGPTL4 mRNA expression was also detected in NAFLD cells. Some reports illustrate that HIFs and hepatic PPARα activity have certain implication in liver toxicity. In addition, we confirmed that PPARα positively bound to HIF-1α promoter. Hence, we next investigated how HIF-1α affects NAFLD through regulating PPARα expression. As shown in Fig. 3A–C, in NAFLD cells, the silence of HIF-1α resulted in the more decline of PPARα and ANGPTL4 mRNA expression. Moreover, the western blot showed that the protein expression of PPARα, ANGPTL4 and HIF-1α in NAFLD cells were also down-regulated by HIF-1α silence (Fig. 3D). PPARα activation increases the antioxidant defense and reduces oxidative stress. Oxidative stress plays an important role in NAFLD because it contributes to the pathological transition of simple hepatic steatosis to steatohepatitis and fibrosis, while the inhibition of oxidative stress is essential to regulate the progression of NAFLD.31 In this study, the content of oxidative stress marker MDA was significantly increased by OA/PA stimulation and further exacerbated by HIF-1α silence (Fig. 3E). MDA can decrease antioxidant enzymes like SOD and cause overproduction of reactive oxygen species (ROS), secretion of proinflammatory cytokines. In fact, accompanying with the more increase of MDA contents, the activities of beneficial antioxidant enzymes SOD and GSH-Px were indeed inhibited by HIF-1α silence in NDFLD cells (Fig. 3F, G). The last decade provided a constellation of findings demonstrating that PPARα behaves as a modulator of both acute and chronic inflammation.32 In this study, the inhibition of PPAR-α/ANGPTL4 singling pathway resulted from HIF-1α silence could significantly promote the secretion of pro-inflammatory IL-6 and TNF-α, reflecting as that the contents of IL-6 and TNF-α in the culture medium of OA/PA-induced HepG2 cells was obviously increased after silencing HIF-1α (P<0.05, Fig. 3H–I). Data above demonstrated that the disruption of HIF-1α could remarkably activate oxidative stress and promote the secretion of pro-inflammatory in NDFLD cells. Collectively, these results suggest that a lack of HIF-1α aggravates lipid deposition in hepatocytes through inhibiting PPAR-α/ANGPTL4 singling pathway.
 PPAR-α, (B) ANGPTL4 and (C) HIF-1α mRNA expression levels were analyzed by qRT-PCR; (D) the protein expression levels of these genes were measured by western blotting. After silencing HIF-1α, MDA concentration (E), SOD and GSH-Px activities (F–G) were detected using the corresponding biochemical test kits. In addition, the secretion levels of IL-1β and TNF-α were also determined by ELISA kits. *P<0.05, **P<0.01 and ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.")
HIF-1α silence-aggravated lipid deposition was closely related to the inhibition of PPAR-α/ANGPTL4 singling pathway. (A) PPAR-α, (B) ANGPTL4 and (C) HIF-1α mRNA expression levels were analyzed by qRT-PCR; (D) the protein expression levels of these genes were measured by western blotting. After silencing HIF-1α, MDA concentration (E), SOD and GSH-Px activities (F–G) were detected using the corresponding biochemical test kits. In addition, the secretion levels of IL-1β and TNF-α were also determined by ELISA kits. *P<0.05, **P<0.01 and ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.
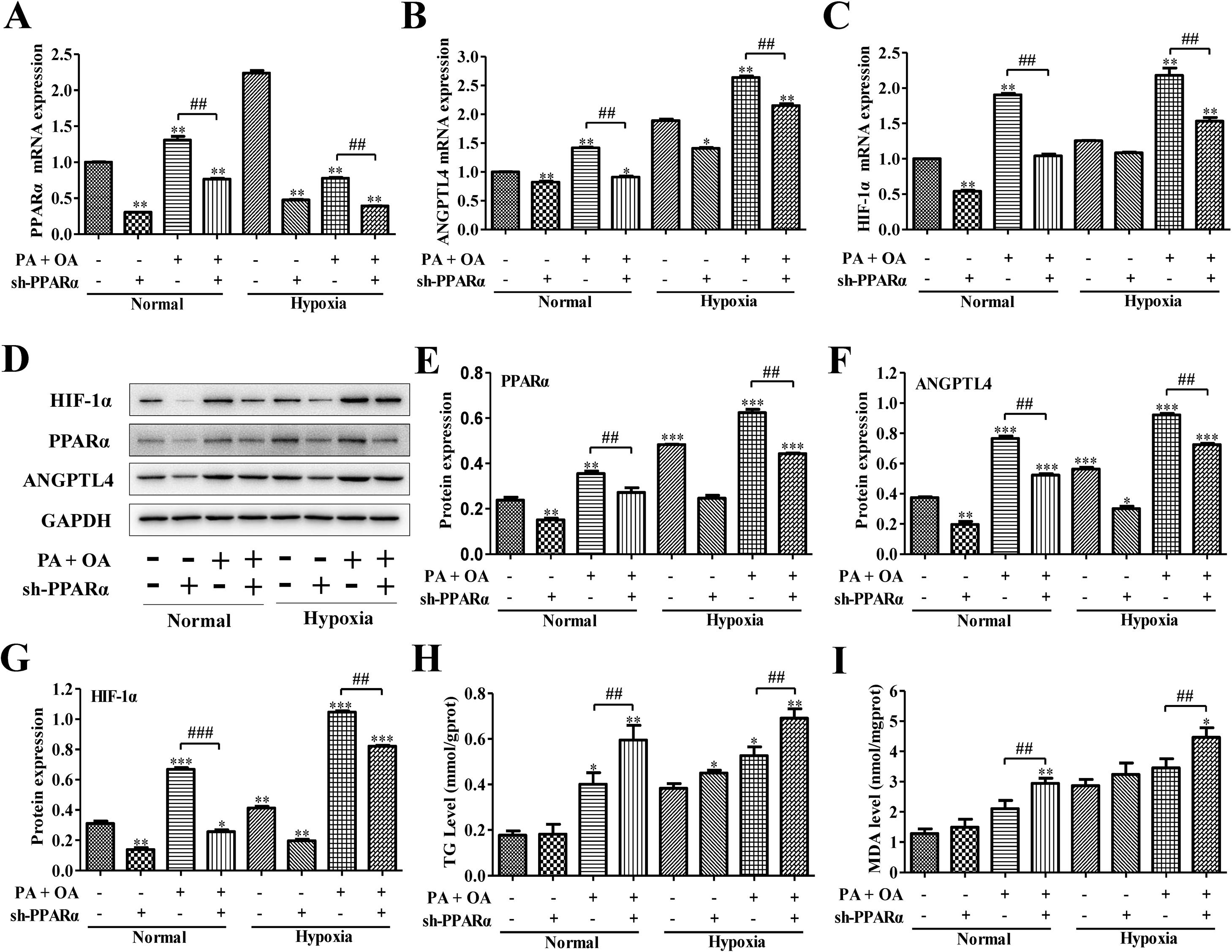
PPARα is an important transcription factor that regulates the expression of genes involved in peroxisomal fatty acid oxidation, and its dysfunction can cause hepatic steatosis.12 We next investigated the role of PPARα in HIF-1α-mediated lipid metabolism in NAFLD cells under normal or hypoxia environment. We utilized adenovirus transfection to silence the PPARα in normal or NAFLD cells on normal or hypoxia environment. Compared with normal environment, hypoxia stimulus significantly enhanced ANGPTL4 and HIF-1α mRNA expression, and partly increased PPARα expression; however, after transfection with sh-PPARα, the mRNA expression levels of PPARα, ANGPTL4 and HIF-1α were inhibited under both normal or hypoxia environment (Fig. 4A–C). Moreover, the expression of PPARα, ANGPTL4 and HIF-1α at protein level were also augmented under hypoxia environment. Once PPARα was silenced by sh-PPARα, the expression levels of these proteins were also obviously down-regulated under both normal and hypoxia environment (Fig. 4D–G). Importantly, under hypoxia environment, the HepG2 cells stimulated by OA/PA without the transfection of sh-PPARα displayed the highest expression of ANGPTL4 and HIF-1α at mRNA and protein levels. In addition, TG and MDA levels were increased in hypoxia, especially under OA/PA stimulation; and it was further aggravated after silencing PPARα (Fig. 4H–I).
 and protein expression levels (D) of PPAR-α, ANGPTL4 and HIF-1α in normal or NAFLD cells with or without sh-PPARα transfection under normal or hypoxic environment were measured and quantified by qRT-PCR and western blot assays, respectively. Moreover, after silencing HIF-1α, the secretion levels of (H) TG and (I) MDA in normal or NAFLD cells under normal or hypoxic environment were measured by biochemical test kits. *P<0.05, **P<0.01 and ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.")
Blocking PPAR-α inhibited HIF-1α-mediated lipid metabolism in NAFLD. The mRNA expression levels (A–C) and protein expression levels (D) of PPAR-α, ANGPTL4 and HIF-1α in normal or NAFLD cells with or without sh-PPARα transfection under normal or hypoxic environment were measured and quantified by qRT-PCR and western blot assays, respectively. Moreover, after silencing HIF-1α, the secretion levels of (H) TG and (I) MDA in normal or NAFLD cells under normal or hypoxic environment were measured by biochemical test kits. *P<0.05, **P<0.01 and ***P<0.001 vs normal cells; #P<0.05, ##P<0.01 and ###P<0.001 vs NAFLD cells without HIF-1α silence.
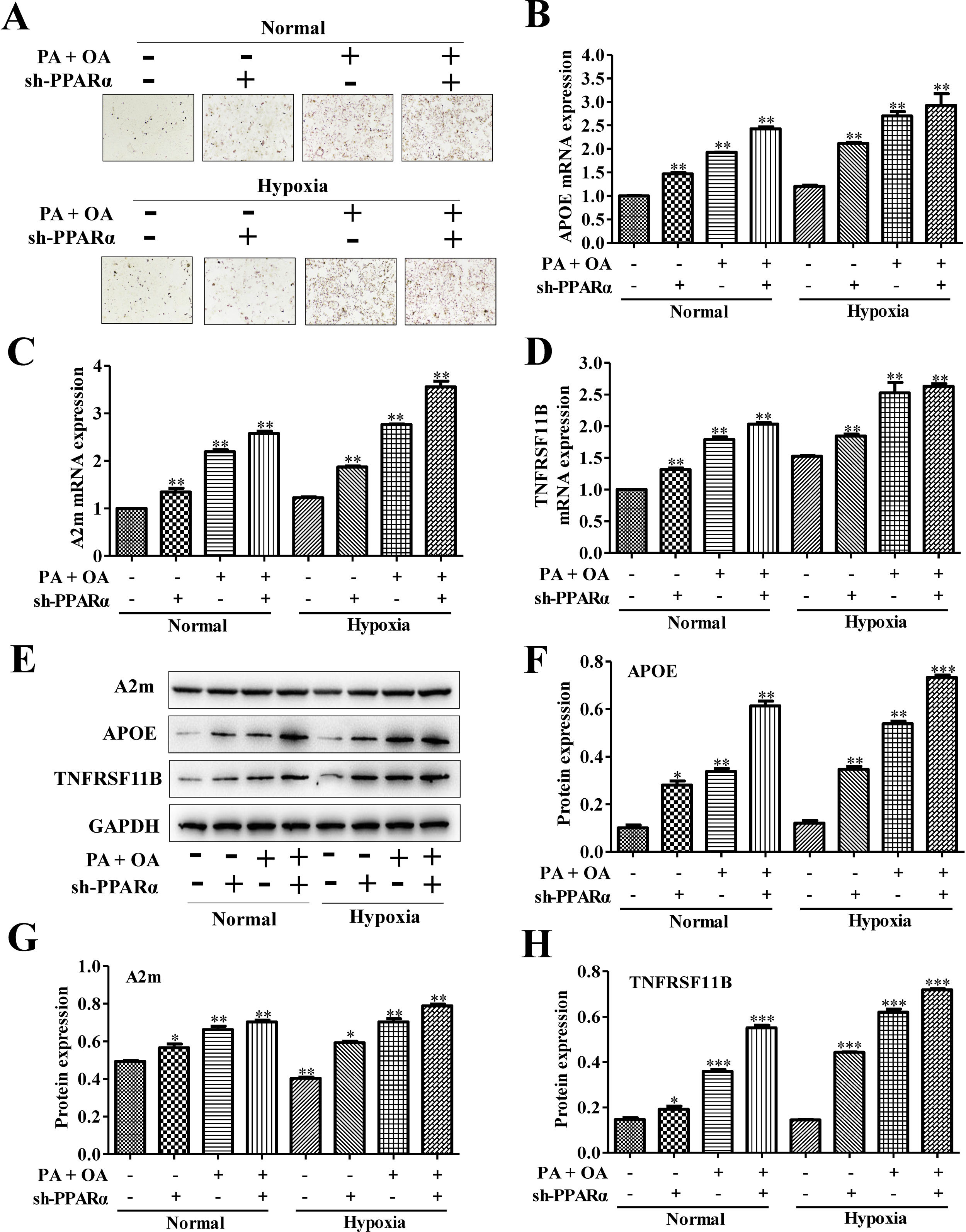
To further investigate the effects of PPARα inhibition on lipid metabolism in NAFLD cells, we used Oil Red O staining to evaluate intracellular lipid accumulation. As shown in Fig. 5A, lipid accumulation was further increased under hypoxia environment. Moreover, the silence of PPARα could aggravate the lipid deposition both under normal and hypoxia environment. Additionally, the mRNA expression of lipid metabolism related genes, including APOE, A2m and TNFRSF11B, were measured, and the results showed that the mRNA expression levels of APOE, A2m and TNFRSF11B could be slightly increased by hypoxia stimulus and be further up-regulated after silencing PPARα (Fig. 5B–D). Similarly, the protein expression of APOE, A2m and TNFRSF11B also exhibited the same tendency (Fig. 5E–H). All of these results illustrated that hypoxia could increase APOE, A2m and TNFRSF11B expression both at protein and mRNA levels and activated PPARα/ANGPTL4 signaling pathway by up-regulating the expression of PPARα and ANGPTL4, blocking PPARα may inhibit PPARα/ANGPTL4 signaling pathway and HIF-1α expression and finally increase the expression of lipid metabolism-related genes and proteins. These suggested that blocking PPARα inhibited HIF-1α-mediated lipid metabolism in NAFLD.
 Oil Red O staining showed that the silence of PPAR-α could exacerbate lipid accumulation in NAFLD cells under both normal or hypoxic environment. The mRNA expression levels (B) and protein expression levels of APOE, A2m and TNFRSF11B in normal or NAFLD cells with or without sh-PPARα transfection under normal or hypoxic environment were detected and quantified using qRT-PCR and western blot assays, respectively. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ###P<0.001 vs NAFLD cells without HIF-1α silence.")
Blocking PPAR-α exacerbated lipid accumulation in NAFLD cells through up-regulating lipid metabolism-related gene. (A) Oil Red O staining showed that the silence of PPAR-α could exacerbate lipid accumulation in NAFLD cells under both normal or hypoxic environment. The mRNA expression levels (B) and protein expression levels of APOE, A2m and TNFRSF11B in normal or NAFLD cells with or without sh-PPARα transfection under normal or hypoxic environment were detected and quantified using qRT-PCR and western blot assays, respectively. **P<0.01, ***P<0.001 vs normal cells; #P<0.05, ###P<0.001 vs NAFLD cells without HIF-1α silence.
NAFLD, an aberrant lipid metabolism disease, has become an increasing prevalent health problem in world.3,33,34 Due to the fatty acid uptake is increased and lipid β-oxidation is impaired, the excessive accumulation of TG in hepatocytes further promotes the NAFLD form.35,36 Much TG stored in hepatocytes forms lipid droplets, and then affects lipid metabolism pathways and leads to the abnormal expression of metabolism-related molecular in NAFLD.35,37 In this study, we utilized HepG2 cells induced by OA and PA to establish in vitro model of NAFLD and found that there was significantly increased intracellular lipid accumulation in NAFLD cells. Consistent with this finding, the contents of TG and ApoB, which are positively correlated with NAFLD incidence, was also elevated in the culture medium of OA/PA-treated HepG2 cells.
HIF includes two distinct subunits oxygen-sensitive HIFα and constitutively expresses HIFβ/aryl hydrocarbon receptor nuclear transcript.38 HIF-1α, a transcription factor, is activated to adapt lower oxygen condition and response to hypoxic.39 NAFLD affects intracellular oxygen diffusion and prevents oxygen delivery through shrinking the blood vessels in liver acinus.40 All of these changed mechanisms and molecules can cause chronic liver hypoxia, and induce different responses hypoxia stress through activating HIF-1α expression in liver. Some reports illustrate that loss of HIF-1α in liver augment hepatic steatosis in mice fed a choline-deficient diet.23 Moreover, the alcoholic fatty liver can be worsened in HIF-1α deficient mice. All of these reports suggest that HIF-1α is a key factor in NAFLD. However, the underlying mechanisms of HIF-1α anti-steatosis effect should be explored. In this study, our results demonstrated that HIF-1α played an important role in inhibiting NAFLD, because it could up-regulate PPAR-α/ANGPTL4 signaling pathway. In addition, we confirmed that the silence of HIF-1α could exacerbate lipid deposition in NAFLD cells through up-regulating the expression of some lipid metabolism-related genes, such as LDLr, ApoE, CPT-1, SREBP-2 and A2m. Hereinto, APOE and LDLr regulates lipid metabolism through regulating lipid transportation and promoting lipid accumulation41; A2m augments glucose uptake, promotes lactate secretion and lipogenesis, and finally aggravates lipid deposition in NAFLD hepatocytes.42CPT-1 regulates β-oxidation of fatty acids; SREBP-2 is closely associated with the lipogenesis. The changes of CPT-1 mRNA expression could cause impaired β-oxidation and lead to lipid accumulation, especially the increase in the contents of LCACoA and TG. In turn, the extensive accumulation of TG in liver cells can induce NAFLD through increasing free fatty acid uptake into the liver. Hence, HIF-1α is a crucial key in NAFLD.
PPARα is a ligand-activated transcription factor, which can bind to fatty acid metabolism gene in tissue.11,12 It can improve steatosis, inflammation and fibrosis in pro-clinical models of NAFLD.12 In addition, we confirmed that PPARα positively bound to HIF-1α promoter. Meanwhile, in our study, a significant decrease of PPARα and ANGPTL4 mRNA expression was also detected in NAFLD cells, which was similar to previous studies that unchanged or even down-regulated PPARα were observed in NAFLD.30 In addition, PPARα activation increases the antioxidant defense and reduces oxidative stress. Here, due to HIF-1α silence-induced downregulation of PPARα and ANGPTL4 mRNA expression, the silence of HIF-1α also resulted in a significant increase in production of oxidative stress marker MDA but a decrease in the activities of beneficial antioxidant enzymes SOD and GSH-Px, suggesting HIF-1α regulates oxidative stress in NDFLD via PPAR-α/ANGPTL4 signaling pathway. Moreover, PPAR-α behaves as a modulator of both acute and chronic inflammation.32 In this study, the inhibition of PPAR-α/ANGPTL4 singling pathway induced by HIF-1α silence could promote the secretion of pro-inflammatory IL-6 and TNF-α, indicating an aggravated inflammation in NDFLD cells. Subsequently, we utilized sh-PPARα to silence PPAR-α and further investigated whether PPAR-α silence affected the expression of lipid metabolism-related genes and proteins. As expected, accompanying by the apparent lipid deposition, the protein and mRNA expression levels of APOE, A2m and TNFRSF11B were all obviously up-regulated in NAFLD cells after silencing PPAR-α both under normal and hypoxic environment, indicating that PPAR-α also regulated the expression of HIF-1α, especially in hypoxia, and it plays an important role in lipid metabolism during NAFLD. In addition, the silence of PPAR-α also led to an excessive increase of TG and MDA secretion.
In conclusion, this study revealed that HIF-1 plays a crucial role in NAFLD. In particularly, HIF-1α could decrease lipid deposition in hepatocytes under OA/PA stimulation through up-regulating PPAR-α/ANGPTL4 signaling pathway and then inhibiting the expression of lipid metabolism-related genes and proteins. It suggests that HIF-1α may become a potential therapeutic target in liver steatosis during NAFLD. However, the effects of HIF-1α on the development of NAFLD should be further investigated, because it is a complicated role in inflammation and tissue remodeling regulation.
Conflicts of interestThere are no conflicts of interest.
This research was financially supported by The Department of Science and Technology of Yunnan Province – Kunming Medical University Joint Research Project (2017FE468-008), The Biomedical Major Special Project of Yunnan Province (2017ZF027 and 2017ZF024-12), The Health Science and Technology Talents (Ten Project) Training Program of Kunming (2016-SW (Country) -5) and The Medical Reserve Talent Development Program of Health and Family Planning Committee of Yunnan Province (H-201645).

