




La enfermedad de Von Hippel-Lindau (EVHL) es un síndrome hereditario poco frecuente que predispone genéticamente a los individuos afectados a la formación de tumores en múltiples sistemas de órganos1,2.
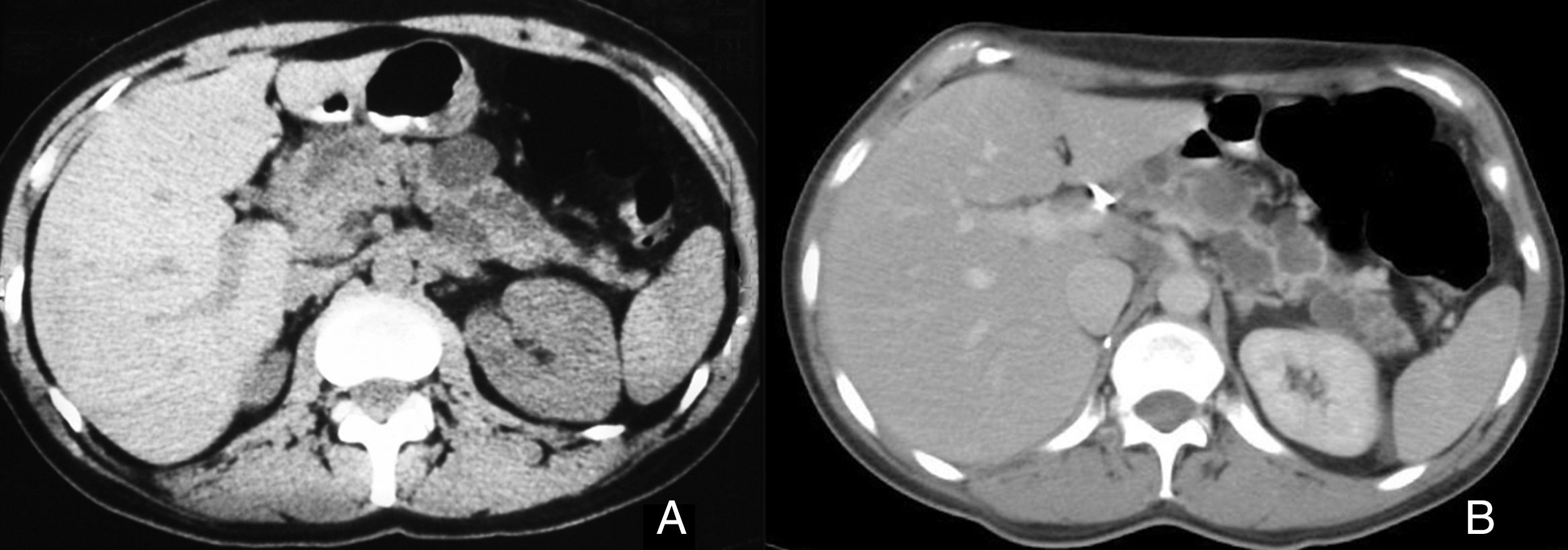
Mujer de 43 años intervenida de un feocromocitoma suprarrenal derecho en 1995. En seguimiento periódico por múltiples quistes simples en la cola del páncreas. En 2007 se aprecia una lesión sólida de 5cm de diámetro a nivel de la cabeza del páncreas que obstruye el conducto de Wirsung y quistes en la cola pancreática (fig. 1A). Analítica sin alteraciones significativas. Se le realiza una duodenopancreatectomía cefálica observando la masa citada en la cabeza pancreática y múltiples quistes en el parénquima pancreático de cuerpo y cola. El postoperatorio cursó sin complicaciones. El estudio histológico fue informado como tumor neuroendocrino de páncreas (TNEP) con positividad a queratinas (AE1/AE3), vimentina, sinaptofisina y enolasa neuronal específica, con índice de proliferación del 18% (MIB-1) y 11 mitosis por 10 campos de gran aumento, además se identificó un cistoadenoma seroso (CAS) y quistes simples de páncreas en el parénquima no tumoral (figs. 2A y B). Ante la sospecha de EVHL se realiza estudio genético que objetiva delección del exón del gen VHL que confirma la enfermedad. La RM craneal descarta alteraciones a nivel de la región hipotálamo-hipofisaria. La paciente tras 10 años de seguimiento presenta un carcinoma renal de células claras pT1a que se extirpa realizando nefrectomía derecha. Los quistes en cuerpo y cola pancreática no han presentado cambios significativos (fig. 1B) y la paciente no presenta insuficiencia pancreática exocrina o endocrina.
 lesión sólida de 5cm de diámetro a nivel de la cabeza del páncreas que obstruye el conducto de Wirsung y quistes en la cola pancreática. B) Quistes en cola pancreática.")
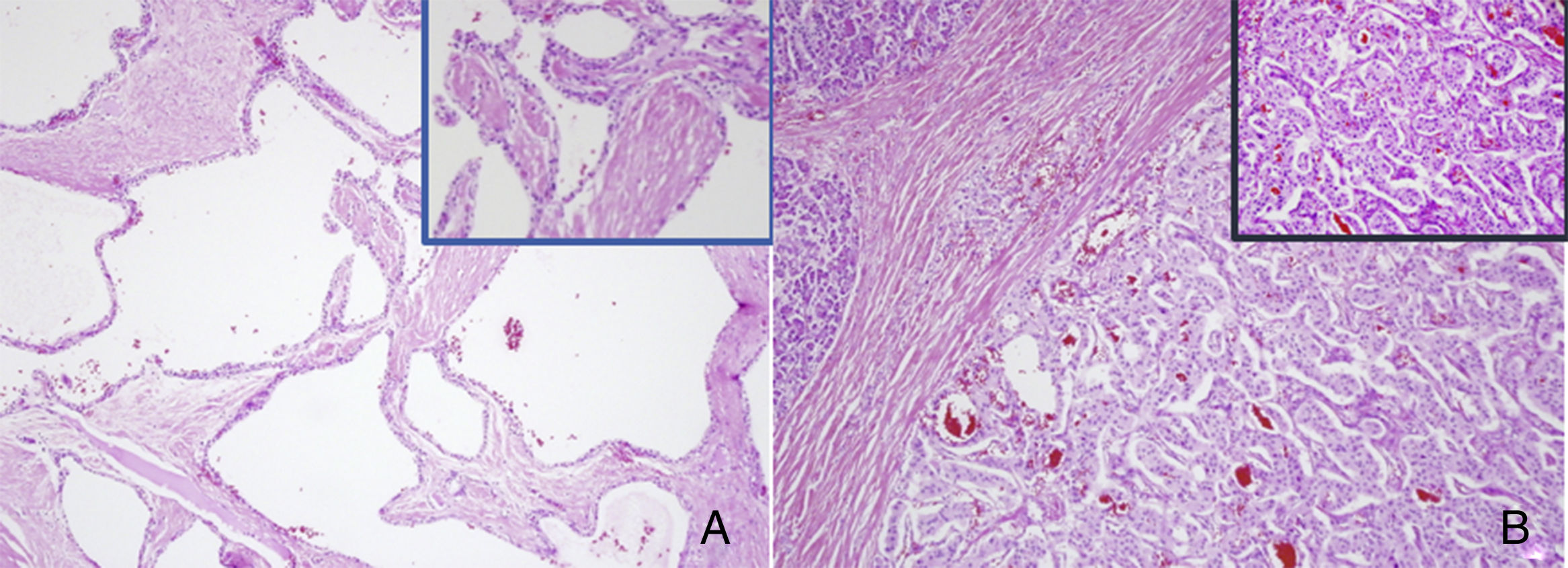
 Cistoadenoma seroso: múltiples estructuras quísticas, recubiertas por epitelio cubico o cilíndrico, con citoplasma claro y núcleo redondeado sin atipia. B) Tumor neuroendocrino: proliferación organoide de células monomorfas, con citoplasma levemente eosinófilo y núcleo ovoideo, regular, con cromatina fina y baja actividad mitótica.")
Estudio histopatológico. A) Cistoadenoma seroso: múltiples estructuras quísticas, recubiertas por epitelio cubico o cilíndrico, con citoplasma claro y núcleo redondeado sin atipia. B) Tumor neuroendocrino: proliferación organoide de células monomorfas, con citoplasma levemente eosinófilo y núcleo ovoideo, regular, con cromatina fina y baja actividad mitótica.
La EVHL es un raro síndrome con herencia autosómica dominante producido por mutaciones en la línea germinal del gen supresor tumoral VHL, localizado en el brazo corto del cromosoma 3 (3p25). Estas mutaciones conducen al desarrollo de tumores benignos o malignos y quistes en diversos órganos, principalmente hemangioblastomas en el sistema nervioso central (SNC) y la retina, carcinomas renales y feocromocitomas3,4. Además, se han asociado otras neoplasias entre las que se incluyen tumores del oído interno, del epidídimo, lesiones quísticas ováricas y diversos tumores pancreáticos3–5. La causa de muerte más frecuente de los pacientes con EVHL son las complicaciones asociadas a los tumores del SNC y el cáncer renal, que implican una expectativa de vida inferior a los 50 años4.
Melmon y Rosen definieron los criterios diagnósticos de EVHL: presencia de un hemangioblastoma en el SNC y otra lesión de EVHL o lesión de EVHL y el antecedente familiar de EVHL. Actualmente las pruebas genéticas se consideran como el estándar para el diagnóstico1.
El riesgo de feocromocitoma se usa para clasificar la EVHL en 2 tipos, este va a depender del tipo de mutación: 1 (riesgo bajo de feocromocitoma pero alto para el resto de los tumores), y 2 (riesgo alto para feocromocitoma) que se subdivide en: 2A: riesgo bajo para tumor renal; 2B: riesgo alto para tumor renal y 2C: solo presentan feocromocitoma3,4,6. Las manifestaciones pancreáticas pueden estar presentes en el tipo 1 (sin feocromocitoma) y en el tipo 2B (con feocromocitoma)7, subtipo que corresponde a nuestro caso por las manifestaciones clínicas. La mayoría de los diagnósticos de EVHL surgen al observar hemangioblastomas en pruebas de imagen realizadas por síntomas neurológicos.
Se estima que entre el 35 y el 70% de los pacientes con EVHL presentan lesiones pancreáticas, a menudo asintomáticas y detectadas incidentalmente1,3,7,8. Generalmente son lesiones quísticas (17-56%): quistes simples o cistoadenomas serosos9 y rara vez una neoplasia endocrina pancreática (10-17%)7,9. Menos frecuentemente (11,5%) se presentan la combinación de estas lesiones como ocurrió en nuestro caso2,3,7.
Las lesiones quísticas del páncreas en la EVHL descritas en la literatura son benignas3,10, pero pueden reemplazar el páncreas y causar insuficiencia pancreática1. Se ha postulado que la sobreproducción del factor de crecimiento endotelial vascular en estos pacientes, puede explicar su alta prevalencia6. Generalmente son quistes múltiples, distribuidos por todo el páncreas, asintomáticos y no funcionales8. Los pacientes precisan de una revisión anual rutinaria mediante tomografía axial computarizada o resonancia magnética para ver su evolución. La indicación quirúrgica solo existe si son sintomáticas y en caso de complicaciones como infección o compresión de órganos o vasos adyacentes3,5. Los cistoadenomas serosos en pacientes con EVHL aparecen típicamente en la tercera década de vida, mientras que en la población general en mujeres en la sexta década10.
Los TNEP en la EVHL, son tumores no funcionantes y de crecimiento lento, y a diferencia de las lesiones quísticas pueden ser malignos y presentar metástasis6. No existen criterios clínicos claros de malignidad, pero se ha postulado el tamaño superior a 2cm y una tasa de duplicación de menos de 500 días4,8,10. El porcentaje de metástasis causadas por los TNEP en EHVL oscila entre el 13 y el 36%3,10, siendo el hígado el sitio más común de metástasis8.
El seguimiento en la EVHL depende del momento y la forma del diagnóstico. En pacientes adultos se realiza TAC abdominal anualmente para el seguimiento y detección de lesiones pancreáticas, así como hepáticas, adrenales y renales2.
El seguimiento de nuestro paciente se realizó con TAC anuales por la presencia de quistes pancreáticos, en este momento se debió sospechar de EVHL y realizar el estudio genético, sin embargo, nuestro diagnóstico fue posterior a la cirugía de una nueva lesión pancreática en cabeza de páncreas con indicación quirúrgica, cuyo estudio histopatológico resulto TNEP y CAS, presentando así las 3 lesiones pancreáticas descritas en la EVHL, confirmándose posteriormente con el estudio genético.
Las lesiones quísticas pancreáticas solo requieren seguimiento y la cirugía se plantea solo en caso de complicaciones, en cambio en los TNEP, la cirugía se plantea por riego de metástasis en función del tamaño.
AutoríasAylhin López Marcano: diseño del estudio, adquisición y recogida de datos, análisis e interpretación de los resultados y redacción del artículo.
José M. Ramia: diseño del estudio, análisis e interpretación de los resultados, redacción del artículo y revisión crítica y aprobación de la versión final.
Roberto de la Plaza: análisis e interpretación de los resultados y revisión crítica y aprobación de la versión final.
Farah Al-Shwely, Alba Manuel Vázquez y Cristina García Amador: adquisición y recogida de datos y análisis e interpretación de los resultados.
Antonio Candia: adquisición y recogida de datos y análisis e interpretación de estudio histopatológico.

