




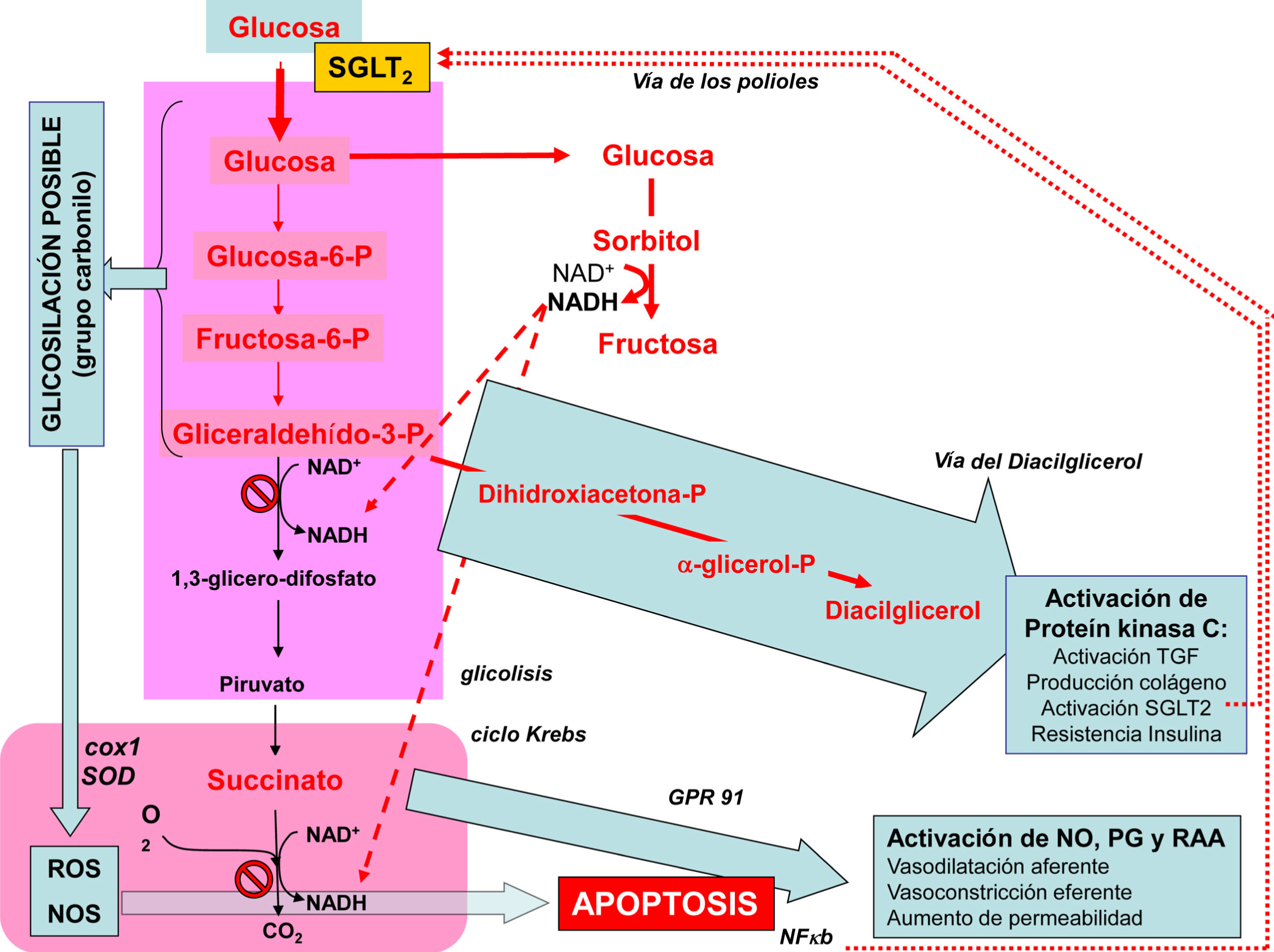
El riñón del diabético presenta un exceso de expresión y actividad del trasportador SGLT2 del túbulo proximal. Esta situación aumenta la reabsorción renal de Na y glucosa, y reduce la oferta distal de los mismos. Además de los efectos metabólicos sobre el medio interno de este exceso de glucosa reabsorbida, el túbulo renal se ve sometido a un estrés glicosilativo capaz de activar localmente tanto apoptosis como inflamasoma. El resultado es la pérdida progresiva de unidades nefronales, la activación de la transición epitelio mensangial y del depósito de colágeno. Se produce la activación de la señalización de insulina por la vía de la MAP kinasa y la resistencia a los efectos metabólicos de la insulina.
Simultáneamente se superpone una vasodilatación aferente por la hiperglucemia, una inhibición del feed-back túbulo-glomerular por la reducción en la oferta distal de fluido, la desdiferenciación de los podocitos y la reducción en su número, estos últimos efectos debidos a la resistencia a la insulina
El resultado es un daño renal autoalimentado, con hiperpresión intraglomerular, desdiferenciación podocitaria, apoptosis tubular y activación local y a distancia del inflamasoma. Todos estos efectos son susceptibles de corregirse total o parcialmente al inhibir el trasporte de glucosa a través de los SGLT2.
The diabetic kidney presents excess expression and activity of the SGLT2 transporter of the proximal tubule. This situation increases the renal reabsorption of Na and glucose and reduces their distal supply. In addition to the metabolic effects on the internal environment of this excess reabsorbed glucose, the renal tubule is subjected to glycosylated stress capable of locally activating both apoptosis and inflammasome. The result is a progressive loss of nephron units, activation of transition of mesangial epithelium and collagen deposition. Activation of insulin signalling by the MAP kinase pathway and resistance to the metabolic effects of insulin take place.
This is simultaneously combined with afferent vasodilation due to hyperglycaemia, tubuloglomerular feedback inhibition due to reduced distal fluid supply, podocyte dedifferentiation and reduction in their number, the latter effects being due to insulin resistance.
The result is self-feeding renal damage, with intraglomerular hyper-pressure, podocyte dedifferentiation, tubular apoptosis, and local and distant activation of inflammasome. All these effects are susceptible to be totally or partially corrected by inhibiting glucose transport via the SGLT transporters.
