




Alkaptonuria is an ultrarare disorder of the amino acid metabolism (Online Mendelian Inheritance in Man [OMIM] number 203500) of autosomal recessive inheritance and with a population frequency between 1/250,000 and 1/1,000,000. It is caused by homogentisate dioxygenase (HGD) mutations leading to an accumulation of homogentisic acid (HGA) that causes darkening of the sclerae and urine, as well as degeneration of the articular cartilage, intervertebral discs, and heart valves.
To date, treatment of this condition has been based on protein restriction to decrease the number of amino acids that can be metabolized to HGA. Vitamin C supplementation supposedly also reduces HGA toxicity. Nitisinone, a hydroxyphenylpyruvate dioxygenase (HPPD) inhibitor that reduces the generation of HGA, has recently been approved for the treatment of this condition, although it tends to increase tyrosine levels1–4.
In this paper we present two cases of alkaptonuria and its successful treatment with low doses of nitisinone.
These cases corresponded to a 57-year-old woman and her 54-year-old brother who were both diagnosed with alkaptonuria due to presenting with dark urine and early degenerative changes in their spine and peripheral joints, owing to which they were referred to our department for follow-up.
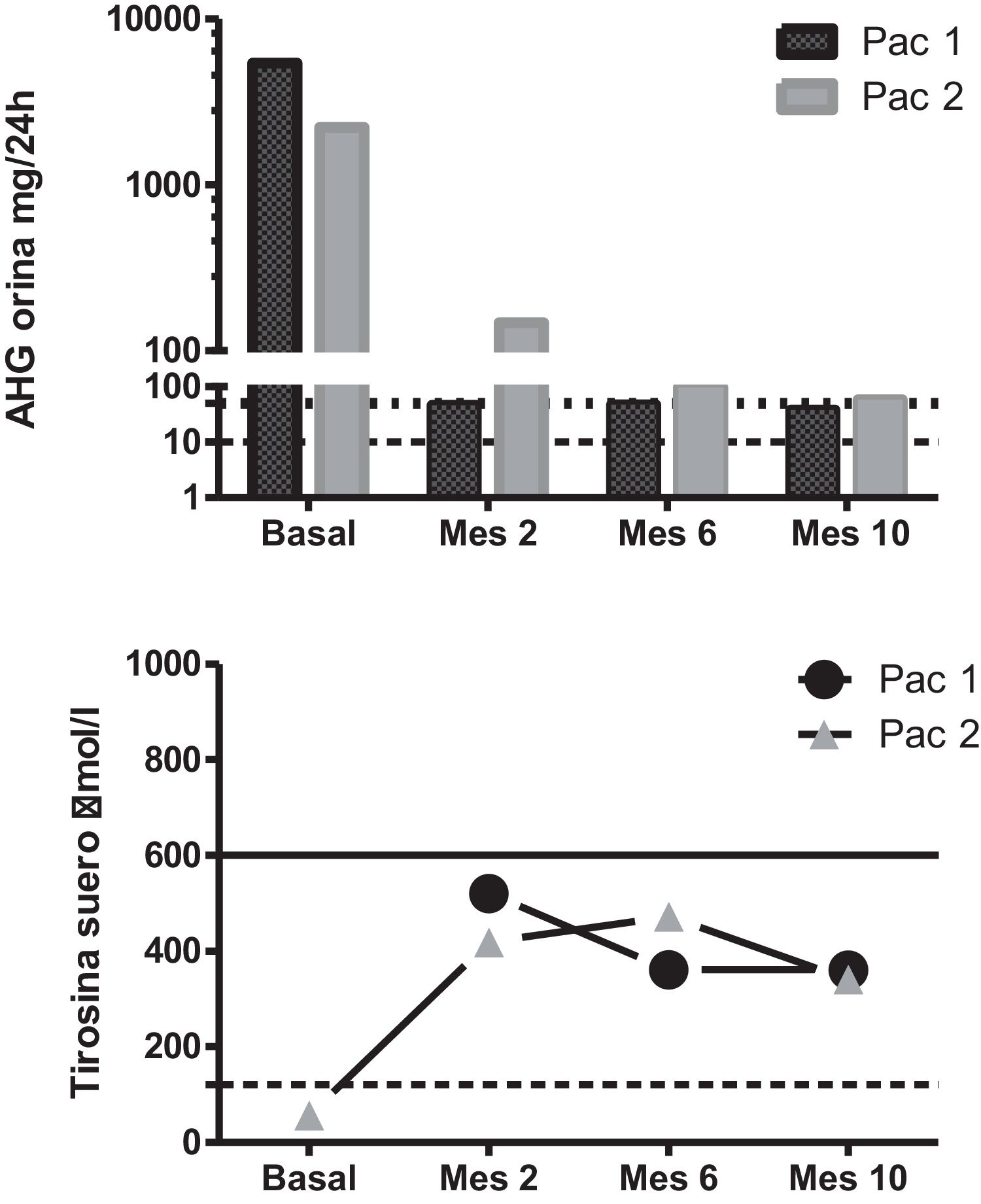
A genetic and biochemical analysis revealed that both patients had double heterozygous mutations in the HGD gene (c.335T > C -p.Phe112Ser- and c.342 + 1G > T) and a high urinary excretion of HGA (Fig. 1). The only child of the female patient was asymptomatic, had a urinary excretion of HGA of 0.8 mg/24 h (reference range 0–10), and also carried mutation c.342 + 1G > T.

Biochemical changes secondary to the treatment. Upper panel: urinary HGA excretion after nitisinone dosing. The dotted line marks the upper limit of the reference range, and the dashed line marks the 50 mg/24 h dosage level. Note the logarithmic scale. Lower panel: serum tyrosine levels after nitisinone dosing. The dotted line marks the upper limit of the reference range, and the dashed line marks the 600 μmol/L threshold.
Pat. 1: patient 1; Pat. 2: patient 2.
Treatment with nitisinone at a dose of 2 mg/day was recommended due to concerns about the side effects associated with the standard 10-mg dose in the context of poor adherence to the required lowprotein diet. Upon starting this treatment, both patients reported an improvement in their musculoskeletal pain, and their urinary HGA decreased by over 97%, to 38 mg/24 h and 58 mg/24 h, respectively. Their tyrosine levels increased to 346–352 μmol/L but remained below 600 μmol/L at all times (Fig. 1).
The c.342 + 1G > T mutation has previously been described in patients with alkaptonuria who were homozygotes or compound heterozygotes. Although we were unable to study the parents of these patients, the genetic and biochemical data of the female patient’s son were consistent with the concept that the patients had mutations in both alleles of the gene, each inherited from one parent. However, to our knowledge, nonsense mutation c.335T > C is new and has not been previously described in the general population or in patients with alkaptonuria. This alteration causes a phenylalanine > serine change affecting a highly preserved residue. This, together with the fact that this mutation was not present in the asymptomatic son, strongly suggests that this is a new pathogenic variant.
Nitisinone was recently approved for the treatment of alkaptonuria based on the results of the SONIA2 study, a controlled clinical trial in which a dose of 10 mg/day was used2. The findings of many studies have shown that nitisinone exerts a dosedependent effect on HGA excretion, but that a dose of 2 mg already decreases HGA levels by more than 90%-95%, as seen in our cases. Even doses as low as 0.2 mg seem to have been effective in some patients3. Although the data yielded by some retrospective analyses suggest that a dose of 10 mg might be more potent in preventing disease progression4, some studies indicate that there may be no differences in the disease’s progression between cases with HGA levels below or above 300 μmol (approximately 50 mg), which is often considered the target lintel of this treatment5.
Ocular and other adverse effects may occur with tyrosine levels >600 μmol/L. These appear to be doserelated, as approximately 15% of patients who received 10 mg of nitisinone and 5% of those who took 2 mg of this drug developed a keratopathy4. Therefore, it remains unclear which dose of nitisinone offers the best risk-benefit ratio5.
In the two patients described in this paper, the low dose of nitisinone provided symptomatic relief associated with a decrease >97% in HGA levels in parallel with an acceptable increase in serum tyrosine levels. We believe that nitisinone is a valuable addition to treatment for alkaptonuria, although the risk-benefit and cost-benefit ratios of the different doses of this drug are still unclear.
Conflicts of interestThe authors of this paper declare no conflict of interest.
