


Introducción
La enfermedad de Parkinson (EP) fue descrita por primera vez en 1817 por James Parkinson, como una condición que consiste en: movimientos temblorosos involuntarios, con disminución de la potencia muscular en la movilidad pasiva y activa, con propensión a encorvar el tronco hacia adelante y de pasar de caminar a correr; los sentidos y el intelecto no sufren mayor daño.1 Actualmente, se define como un desorden neurológico progresivo que surge como resultado de la pérdida de células dopaminérgicas en la parte compacta de la sustancia negra.
Además de la sintomatología previamente descrita, la EP también se acompaña de temblor en reposo, rigidez, bradicinesia e inestabilidad postural.2 También puede ser acompañada por manifestaciones psiquiátricas como depresión y alucinaciones visuales. Los síntomas no motores, como pérdida del olfato, síntomas autonómicos y demencia, afectan la calidad de vida y esperanza de vida de estos pacientes. Los síntomas no motores y psiquiátricos no están presentes de forma uniforme en todos los pacientes. La demencia afecta 20% a 40% de los casos. La variabilidad clínica se debe a la influencia de múltiples factores ambientales y genes de susceptibilidad.
La EP es la forma más frecuente de parkinsonismo y el segundo desorden neurodegenerativo más frecuente, después de la enfermedad de Alzheimer.3 Se estima una prevalencia de 0.3% en la población general. Cabe recalcar que de 1% a 3% de todas las personas mayores de 60 años padece EP. Como la esperanza de vida ha ido en aumento, se considera una patología importante y frecuente en la población adulta mayor. La incidencia general es de 13.4 por 100 000 personas.
Desde el punto de vista genético, es una enfermedad compleja y multifactorial ya que contribuyen factores genéticos y ambientales. Hasta ahora, las causas genéticas han podido explicar hasta 10% de todos los casos de EP. El análisis de los antecedentes familiares permite dividir a la EP en esporádica y familiar.3 De los casos, 10% a 25% presentan un patrón de herencia familiar determinado, ya sea dominante o recesivo. El resto de los casos, en donde no se puede identificar una mutación o que no presenta un patrón de herencia específico, se consideran esporádicos. Como la EP es una enfermedad genéticamente heterogénea y de herencia compleja el riesgo empírico para cualquier persona a desarrollar EP es de 1% a 2%. Cuando hay antecedentes familiares positivos para esta enfermedad el riesgo acumulado para desarrollar EP es de 3% a 7% haciendo notar la importancia de los factores genéticos asociados.
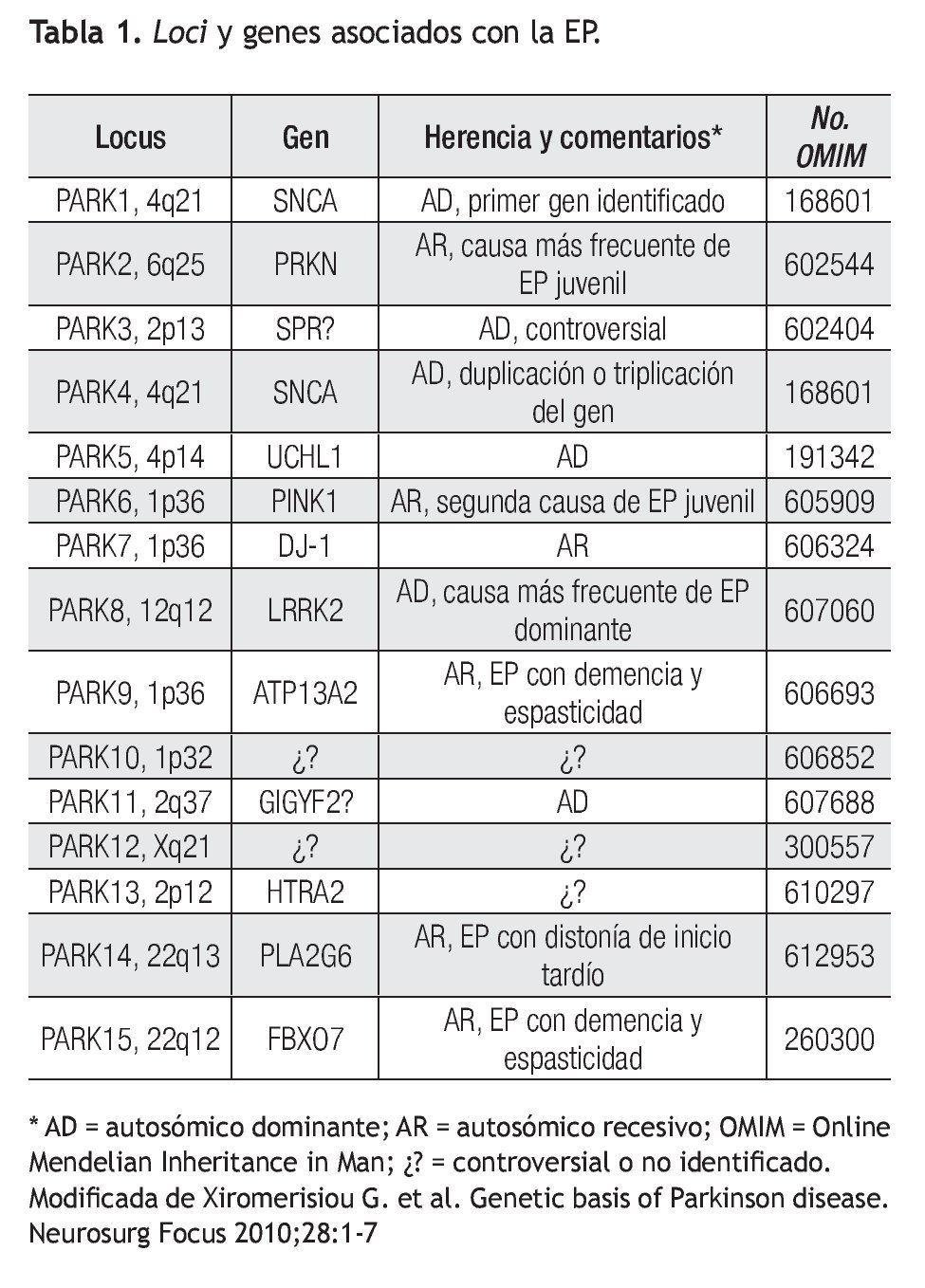
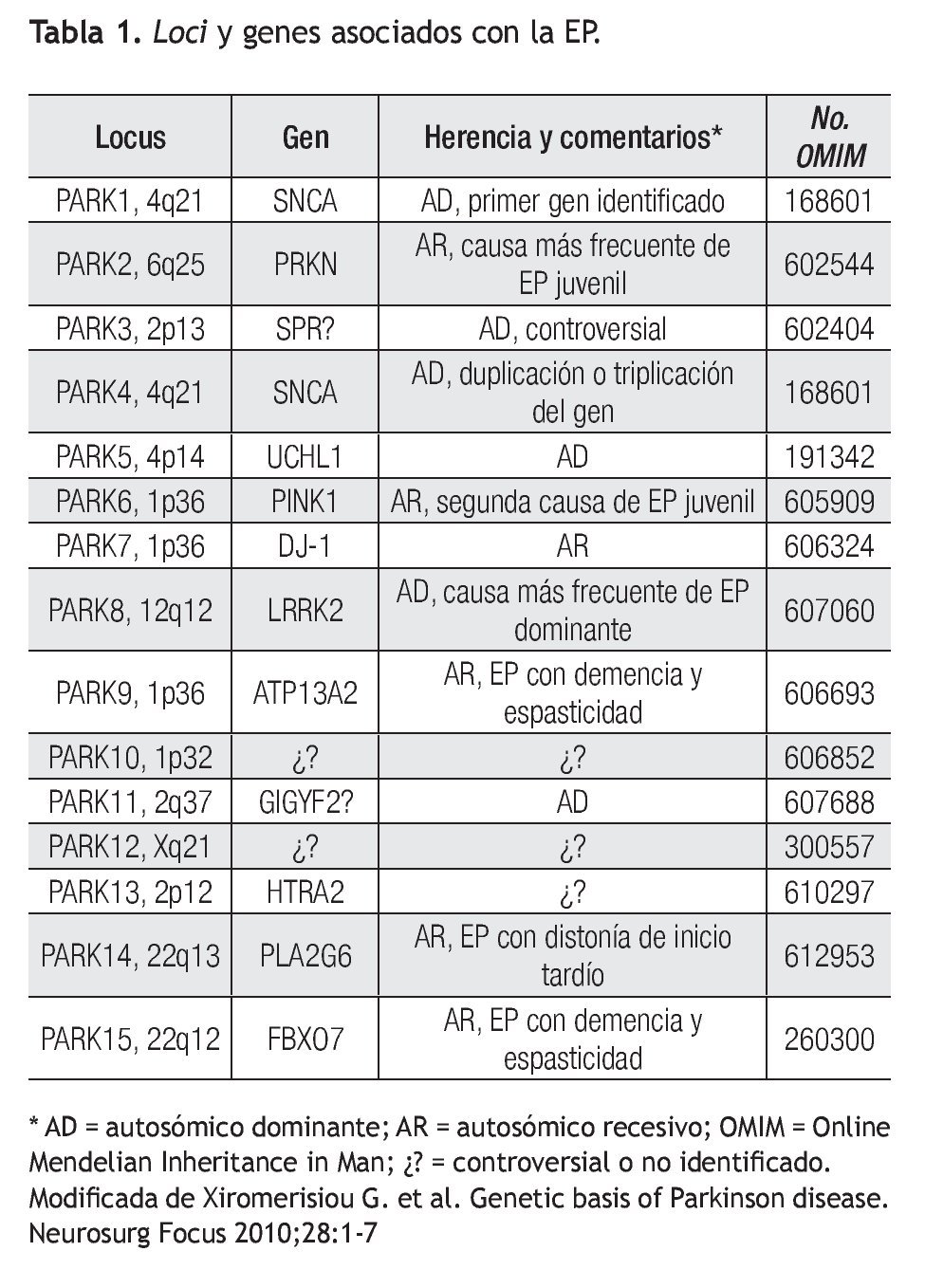
Se han identificado mutaciones y polimorfismos en más de 10 genes relacionados con la EP ya sea como causales o genes de susceptibilidad para la enfermedad.4 Como por ejemplo, la presencia del alelo ε4 de ApoE, al igual que en la enfermedad de Alzheimer, ha sido asociado como factor de susceptibilidad para desarrollar EP.5 Seis de estos genes (PARK1, PARK2, PARK5, PARK6, PARK7 y PARK8) han sido fuertemente implicados como causales o como factores de susceptibilidad para EP.3-5 PARK3 sólo se ha identificado en familias alemanas y aunque el locus ha sido mapeado a la región 2p13, no se ha podido identificar el gen responsable. El gen SPR que codifica para la sepiapterina reductasa es considerado como gen candidato para PARK3 ya que se ha demostrado asociación con EP esporádico y familiar.6 PARK4 ya no se considera como un locus independiente ya que se demostró ser una expansión del gen de PARK1 (α-sinucleína). Existen otros locus recientemente relacionados con EP pero aún no se ha podido establecer su significancia clínica, por lo cual continúan en investigación.7
Es importante recalcar que las mutaciones reportadas en PARK1, PARK2, PARK5, PARK6 y PARK7 explican hasta 5% de todos los casos de EP. PARK8 o LRRK2 explica 2% a 7% de todos los casos de EP a nivel mundial. Este último, también ha demostrado ser responsable de 20% a 40% de los casos de EP en judíos Askenazi y árabes del norte de África.8 Es por eso que el análisis de polimorfismos y mutaciones del gen LRRK2 se considera un factor genético importante ya sea como factor de riesgo a desarrollar la enfermedad o hasta como un gen de causalidad con herencia autosómica dominante. Por lo tanto, la búsqueda de otros factores de riesgo genéticos de susceptibilidad continúa en vigor.
La enfermedad de Parkinson (EP) presenta un cuadro clínico característico conformado por temblor en reposo, rigidez, bradicinesia e inestabilidad postural. Cabe recalcar que se acompaña de muchos otros síntomas no motores que coexisten con los motores. Hasta 60% de los pacientes presentan síntomas adicionales que consisten en alteraciones autonómicas, sensoriales y dificultades cognitivas y del comportamiento. Por ejemplo, la depresión y la demencia contribuyen a un descenso en la calidad de vida independientemente de las dificultades motoras.
El manejo y tratamiento de estos síntomas asociados son de igual importancia que los síntomas motores ya que permiten un mejor manejo y control del paciente y su calidad de vida. Aunado a esto se asocia la percepción de la EP en los familiares del paciente y los riesgos que pudieran existir de que ellos manifiesten el padecimiento. Es por eso que se requiere un equipo multidisciplinario para el apoyo y manejo integral del paciente y la familia con EP.
El asesoramiento genético es de igual importancia que el manejo sintomático del paciente con EP ya que se puede establecer un patrón de herencia y determinar factores genéticos que pudieran agregar susceptibilidad a presentar la enfermedad. La investigación genética molecular ha dedicado tiempo en detectar genes responsables de la EP y lograr establecer una correlación genotipo-fenotipo.9
La EP se considera esporádica en la gran mayoría de los casos (casi 90%) pero estudios de ligamiento han permitido identificar genes responsables de formas familiares de EP con patrón de herencia autosómico dominante y recesivo. Aproximadamente, sólo 5% a 10% de todos los pacientes con EP son portadores de una mutación conocida responsable de causas monogénicas de este desorden.10 En la Tabla 1 se puede observar los genes y loci relacionados hasta el momento con la EP. A continuación se describen las formas monogénicas de la EP con alta relevancia a nivel mundial en un orden cronológico y posteriormente, se describen otros genes y loci implicados con la EP pero con una colaboración incierta o controversial.
Formas monogénicas de la Enfermedad de Parkinson
El gen PARK1/PARK4 o SNCA codifica para la α-sinucleína, que ha sido fuertemente asociada a EP familiar formando cúmulos de proteína anormal conocidos como cuerpos de Lewy.4 Han sido identificados previamente en casos familiares de EP con herencia autosómica dominante tres mutaciones de sentido equivocado (A53T, A30P y E64K) así como duplicaciones y triplicaciones de este locus que contiene al gen SNCA. El fenotipo de estos pacientes se caracteriza por ser EP de inicio temprano, progresión rápida y alta prevalencia de demencia y otras alteraciones psiquiátricas y autonómicas. Se cree que los cambios en la expresión o la presencia de mutaciones en la α-sinucleína tienen efectos neurotóxicos en las neuronas dopaminérgicas. Los monómeros de α-sinucleína forman fibrillas y protofibrillas que son tóxicas en las neuronas productoras de dopamina en la sustancia negra.
El gen PARK2 o PRKN codifica para la proteína parkina, con mutaciones asociadas a formas autosómicas recesivas de inicio temprano o juvenil de EP con progresión lenta.10 Se han descrito múltiples mutaciones en este gen responsables de la forma autosómica recesiva. También se han reportado polimorfismos como S167N, R366W y V380L asociados a un riesgo elevado de presentar EP esporádico.11 La parkina se localiza de forma preferencial en las sinapsis y su principal función es de ubiquitin ligasa, importante en la vía de degradación proteín-ubiquitina. Se caracteriza por presentar degeneración en la parte compacta de la sustancia negra con ausencia de cuerpos de Lewy.
PARK6 o PINK1 codifica para la proteína cinasa PTEN 1 (PTEN induced putative kinase 1) y el gen PARK7 o DJ-1 codifica para la proteína DJ-1, ambas implicadas en procesos de neuroprotección. Es decir, la alteración en estas proteínas está implicada en disfunción mitocondrial y estrés oxidativo. También, ambos genes han sido mapeados en 1p36. Aunque aún no se comprende la patología, se han encontrado mutaciones en ambos genes en pacientes con EP de inicio temprano pero con progresión característicamente lenta y alta incidencia de manifestaciones psiquiátricas.12 La incidencia de estas alteraciones es desconocida pero se considera rara, responsable de 1% de los casos de EP autosómico recesivo de inicio temprano.9
De manera particular, el gen PARK8 o LRRK2 codifica para la proteína dardarina o LRRK2 (leucine-rich repeat kinase 2) la cual posee una actividad de cinasa que permite interactuar con la proteína parkina. Su disfunción se asocia a una incapacidad de mantener la estabilidad de las neuronas dopaminérgicas.7,13-15 También se ha asociado a una disfunción en los mecanismos de transporte vesicular intracelular, es decir en la regulación de la endocitosis de vesículas sinápticas.16
Se han descrito más de 40 mutaciones en donde la gran mayoría son mutaciones de sentido equivocado. La mutación G2019S del gen LRRK2 es la responsable de 1% a 2% de los casos esporádicos de EP y de 5% a 6% de los casos familiares de EP en árabes de África del Norte y con una alta prevalencia en judíos askenazi (20% a 40%).8,17 Esta mutación ha mostrado ser de penetrancia incompleta (sólo 32%). Esta mutación no se ha reportado en la población asiática, y es considerada como la más frecuente en LRRK2.18,19 La segunda mutación más importante reportada en el gen LRRK2 es la R1441G/C, que aunque se desconoce su prevalencia a nivel mundial, parece estar restringida a pacientes con ascendencia española o hispana, sobre todo de la etnia vasca.12 Como se sabe, la población mexicana está compuesta por un alto mestizaje con ascendencia indígena, asiática y española, incluida la etnia vasca. La tendencia actual es que LRRK2 se comienza a considerar como el principal gen relacionado no sólo con la EP con herencia autosómica dominante, sino también con la EP de tipo esporádico.
Otros genes y loci asociados con la Enfermedad de Parkinson
El gen PARK5 o UCHL1 codifica para una proteína de la familia de ubiquitinas conocida como esterasa ubiquitina carboxi-terminal L1 asociada a EP. La mutación I93M del gen UCHL1 sólo se ha encontrado en una familia de origen alemán. En múltiples estudios posteriores, no se ha podido identificar otros portadores de esta mutación u otras mutaciones, por lo tanto, este reporte pudo ser el resultado de un polimorfismo coincidental, más que ser considerado como factor de riesgo para EP.3,6,12
El gen ATP13A2 mapeado en el locus de PARK9 es responsable de una forma atípica de parkinsonismo conocida como enfermedad de Kufor-Rakeb.8 Se caracteriza por presentar un patrón de herencia autosómico recesivo, ser inicio juvenil con degeneración piramidal y disfunción cognitiva. El gen ATP13A2 codifica para una proteína lisosomal tipo-P involucrada en las vías de degradación celular. Se cree importante para la degradación de agregados de α-sinucleína y que su disfunción podría explicar los síntomas de parkinsonismo.20 Se debate si esta patología debe ser considerada como un fenotipo diferente y aparte de la EP clásica.
El gen GBA codifica para la glucocerebrocidasa y cuya versión mutante es la causa de enfermedad de Gaucher, una enfermedad lisosomal autosómica recesiva. Mutaciones en el gen GBA también han sido fuertemente asociadas con la EP. Se han reportado más de 200 mutaciones y se estima que 2% a 4% de los pacientes con EP caucásicos presentan mutaciones en GBA.21 Se cree que estas mutaciones actúan como factor de riesgo para presentar la EP.
Otros loci han sido asociados como candidatos pero los genes responsables aún no han sido identificados, como es en el caso del locus PARK12 mapeado en Xq21. En otros casos, la significancia clínica de ciertos loci no han sido demostrada (PARK3, PARK10, PARK11 y PARK13). En el caso del gen PLA2G6 o PARK14 y del gen FBXO7 o PARK15, que participan en vías de degradación ubiquitinproteosoma, han sido asociados únicamente a formas atípicas de EP.
Conclusión
Siendo la EP la segunda enfermedad neurodegenerativa más frecuente que afecta a la población mundial, es comprensible que la investigación médica clínica invierta recursos para elucidar los misterios que rodean a esta enfermedad progresiva. Poco a poco, el manejo y tratamiento de la EP se va convirtiendo en un trabajo multidisciplinario. Como parte de ese equipo se encuentra la genética médica, en donde trata de explicar factores genéticos hereditarios y cómo estos factores influyen en la manifestación clínica de la EP.
Como se explicó con anterioridad, la EP es una enfermedad compleja multifactorial en donde no sólo los factores ambientales son importantes. Tampoco se trata de una enfermedad monogénica en donde un solo gen es responsable de la enfermedad. Hasta ahora, existen más de 10 loci asociados a la EP sin contar muchos otros factores genéticos que han sido implicados como factores de susceptibilidad que elevan el riesgo a presentar dicha enfermedad. Esto es de gran importancia ya que permite al equipo multidisciplinario comprender mejor la enfermedad y basar el manejo y tratamiento específico para cada paciente y su familia.
El conocer el gen responsable de la EP en una familia permite una atención personalizada, brindar el asesoramiento genético correcto con riesgos de recurrencia específicos para esa familia. Cabe recalcar que aunque existe la posibilidad de realizar el diagnóstico molecular, sigue siendo un tema de gran controversia el realizar este estudio a familiares no afectados debido a las implicaciones éticas y personales que esta información pueda condicionar. También es importante señalar que la penetrancia no es completa, esto quiere decir que aunque una persona sea portadora de una mutación, no garantiza que manifestará la enfermedad y por lo tanto, el efectuar éstas pruebas en personas asintomáticas debe realizarse con cautela.
El identificar una causa genética de la EP permitirá ofrecer a estos pacientes y sus familias una herramienta potencialmente útil: terapia génica. Actualmente, existen cuatro terapias génicas en estudio clínico en fase I/ II para tratar los síntomas motores de la EP.22 La terapia génica consiste en la inyección directa de vectores virales no tóxicos con altos niveles de transducción génica. Una de las terapias en estudio está dirigida a favorecer la expresión de genes neuroprotectores para evitar la muerte neuronal. Otras terapias se han enfocado en aumentar la producción endógena de dopamina, mientras otras terapias consisten en controlar vías alternas a la dopamina para eliminar los síntomas motores asociados a la pérdida de las neuronas de la sustancia negra.
Aún con todos los avances, la genética sólo explica 10% de todos los casos de Parkinson. Es decir, más de 90% de los casos de Parkinson no pueden ser explicados por los conocimientos actuales de la medicina asociada a EP. La terapia génica se propone como una herramienta potencial para tratar los síntomas motores para ese bajo porcentaje de pacientes en donde una causa genética ha sido identificada.21 Existe aún mucho campo de investigación en genética y en otras ramas de la medicina relacionado a esta enfermedad neurodegenerativa, con la esperanza de que en poco tiempo se comprenda aún más la patogenia de la EP y poder abocar la fuerza médica a contrarrestar los síntomas neurodegenerativos y mejorar la calidad de vida de estos pacientes.
Correspondencia: Gabriela Elizondo Cárdenas.
Ave. Francisco I. Madero y Dr. Eduardo Aguirre Pequeño, SN Col. Mitras Centro, Monterrey, Nuevo León, México CP: 64460.
Teléfono/Fax: (81) 8329 4217 y (81) 8348 3509.
Correo electrónico:gabrielaec@gmail.com
Recibido: Enero 2011.
Aceptado: Abril 2011