



Nonketotic hyperglycinaemia (NKH), also known as glycine encephalopathy (MIM #605899), is an inborn error of metabolism affecting the mitochondrial glycine cleavage system, and follows an autosomal recessive inheritance pattern.1,2 The condition results in glycine accumulation, which stimulated N-methyl-D-aspartate (NMDA) receptors, causing most of the symptoms.3 NKH has several clinical forms. The most frequent is the classic form, characterised by hypotonia, abolished Moro reflex, myoclonic seizures, apnoea, lethargy, and coma; symptoms appear in the first days after birth.4 Other less frequent variants have been described, including late-onset NKH5 and 3 atypical forms with more heterogeneous symptoms.1,6
During the first month of life, EEG displays a burst-suppression pattern,4 which later changes to hypsarrhythmia. Biochemical diagnosis is based on quantification of glycine levels in the plasma/serum and in the CSF, combined with urine organic acid analysis to rule out organic aciduria.3 CSF/plasma glycine levels are higher than 0.08 in the classic type of NKH and range from 0.04 to 0.02 in the atypical forms.3,5 However, some authors have found no correlation between symptoms and CSF/plasma glycine levels.7,8 Treatment of NKH consists in limiting protein intake, combined with the administration of sodium benzoate. Patients may also receive NMDA receptor antagonists.9 Valproate is contraindicated as it increases glycine levels.10,11
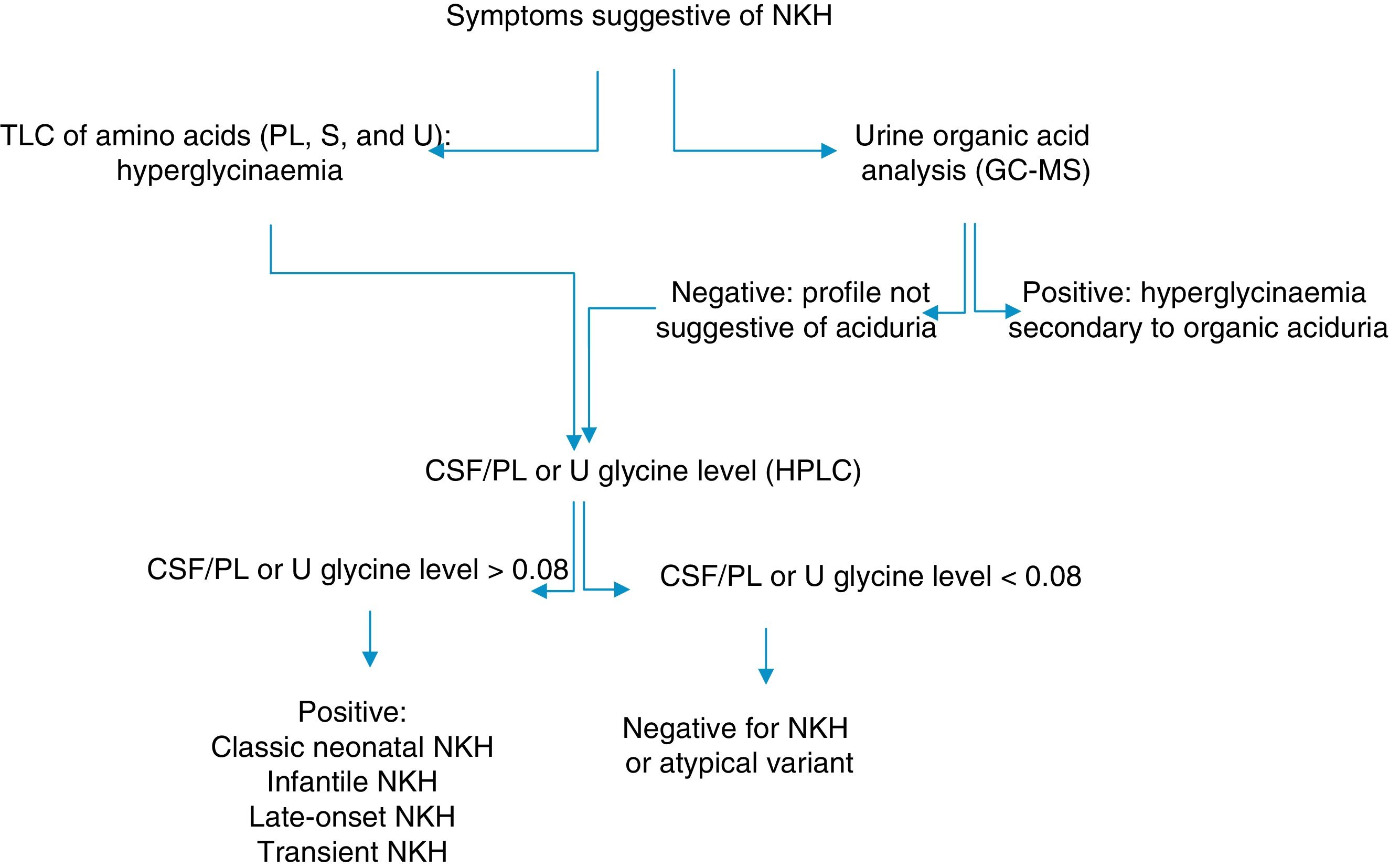
In Cuba, organic aciduria is frequently suspected in patients with NKH. NKH should therefore be ruled out in patients with hypotonia, encephalopathy, and seizures without a diagnosis of an inborn error of metabolism. We describe the clinical and biochemical profiles of 2 patients displaying symptoms of NKH and provide the algorithm used for biochemical diagnosis (Fig. 1).

Algorithm for the biochemical diagnosis of nonketotic hyperglycinaemia.
CSF: cerebrospinal fluid; GC–MS: gas chromatography–mass spectrometry; HPLC: high-performance liquid chromatography; NKH: nonketotic hyperglycinaemia; PL: plasma; S: serum; TLC: thin-layer chromatography; U: urine.
Our first patient began to display neurological symptoms when she was 28 days old: hypotonia, lethargy, apnoea, and myoclonic seizures. Suspicion of West syndrome was confirmed by EEG findings (burst-suppression during the first weeks of life, progressing to hypsarrhythmia a month after birth). The patient was initially treated with valproate and gabapentin (vigabatrin 60mg/kg/day), with no improvement. Seizures resolved with adrenocorticotropic hormone and topiramate. Laboratory analysis revealed a glycine level of 109μM in the CSF and 351μM in the serum (CSF/serum ratio, 0.3 [normal range, <0.08]). Analyses performed a month later revealed a glycine level of 366μM in the serum and 130μM in the CSF (CSF/serum ratio, 0.4). The patient was diagnosed with classic neonatal NKH.
Patient 2The second patient began to exhibit symptoms suggestive of an inborn error of metabolism at the age of one year. The main symptoms were refractory generalised epilepsy, intellectual disability, and motor developmental delay; EEG findings were pathological. The patient received sodium valproate (750mg/day) and clobazam (25mg/day). He had a glycine level of 102μM in the CSF and 88μM in the serum (CSF/serum ratio, 1.2). These results, combined with the patient's symptoms, suggested infantile NKH or valproate-induced NKH. Glycine levels decreased to 38μM in the CSF and 112μM in the serum following withdrawal of valproate. The CSF/serum ratio was 0.3, however, and the patient was diagnosed with infantile NKH.
In Cuba, biochemical screening for NKH is performed only in symptomatic patients; the newborn screening programme does not include this condition. CSF glycine quantification should be performed in patients with high urine and blood glycine levels in whom organic aciduria has been ruled out. The patients’ parents and relatives were advised about the 25% risk of recurrence, given the autosomal recessive inheritance pattern of the condition. Prognosis was based on the patients’ age and initial symptoms, as no molecular characterisation of the disease was performed.
Determination of CSF/serum glycine levels is essential in the diagnosis of the classic type and most atypical forms of NKH.1,5,12,13 Complementary tests are recommended to confirm the diagnosis of NKH in patients with positive results or who may have an atypical form of the disease with a normal CSF/serum glycine ratio.
Please cite this article as: Contreras-Roura J, Camayd-Viera I, Padrón-Díaz AD, Martínez-Rey L. Diagnóstico bioquímico de la hiperglicinemia no cetósica en Cuba. Neurología. 2018;33:549–550.
