



Creutzfeldt-Jakob disease (CJD) is the most frequent type of prion disease in humans, resulting from a conformational change from the normal cellular prion protein (PrPC) to a pathological prion protein (PrPSc), with aggregates in the central nervous system.1 CJD may be classified aetiologically as infectious/acquired, sporadic (SCJD), or familial (FCJD).2 Familial forms of CJD are caused by mutations in the codification of PrP genes; the largest group of patients affected by FCJD is found among Jews of Libyan and Tunisian descent who are carriers of the E200K mutation (Glu to Lys substitution) in the PRNP gene.3,4 Between 1997 and 2008, a reference centre for CJD established that 15.6% of 517 patients analysed carried the E200K mutation.5 Definitive diagnosis of CJD is based on the histological identification of spongiform degeneration in tissues of the central nervous system. Diagnosis of probable CJD is based on clinical findings, typical electroencephalography (EEG) readings, and/or presence of 14.3.3 protein in the cerebrospinal fluid together with a history of the disease of under 2 years’ duration.2 In the early phases of the disease, EEG may be normal or may display non-specific changes, such as delta or theta activity. Occasionally, the first EEG finding may be frontal intermittent rhythmic delta activity (FIRDA) or periodic lateralised epileptiform discharges (PLED). Several weeks later, EEG may reveal periodic sharp wave complexes (PSWC) with a frequency of 1–2Hz.6 In patients with an E200K mutation, sleep disorders are a frequent first manifestation, with video-polysomnography (V-PSG) studies revealing an abnormal sleep architecture, multiple jerking movements,7 and central and obstructive apnoea.1
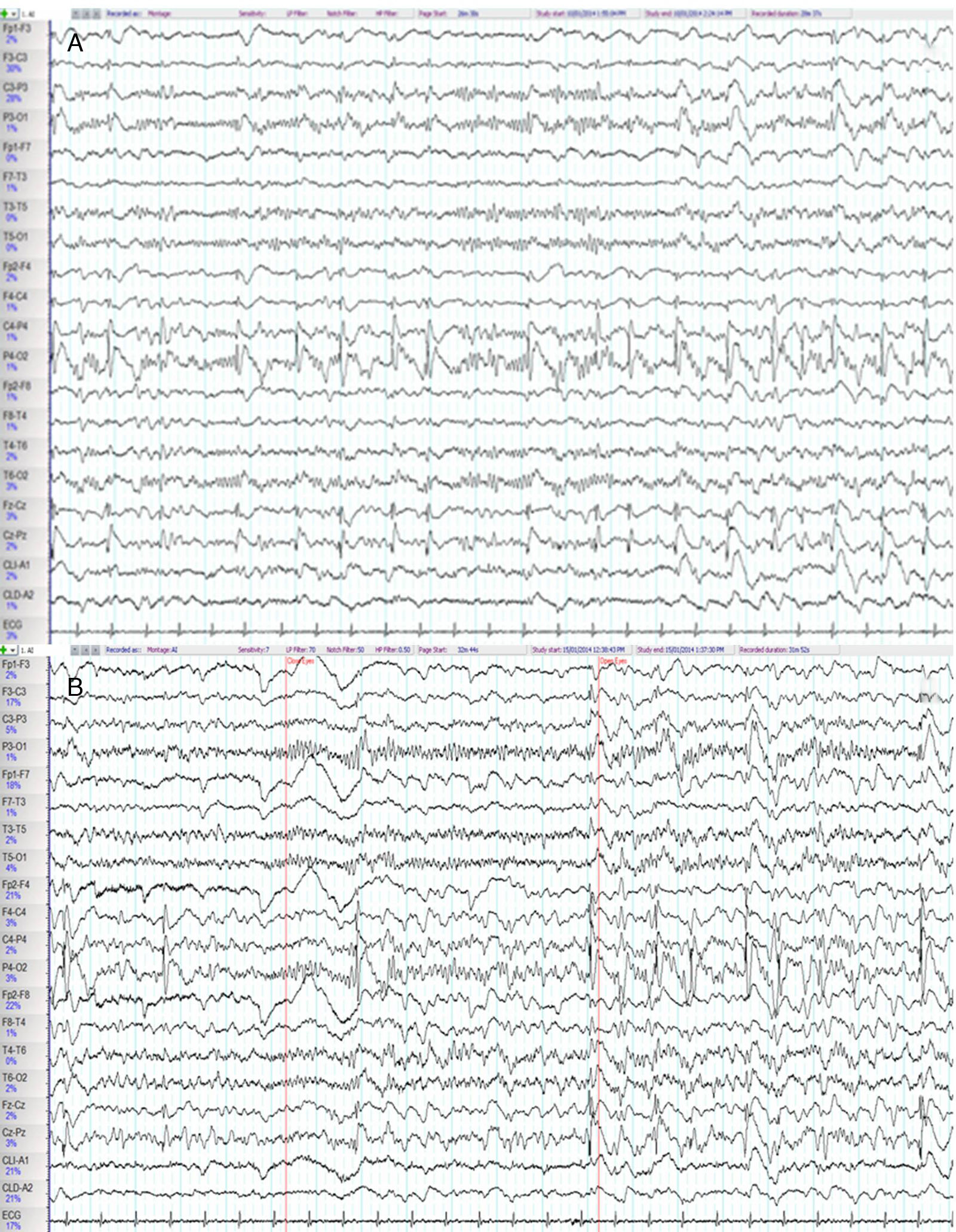
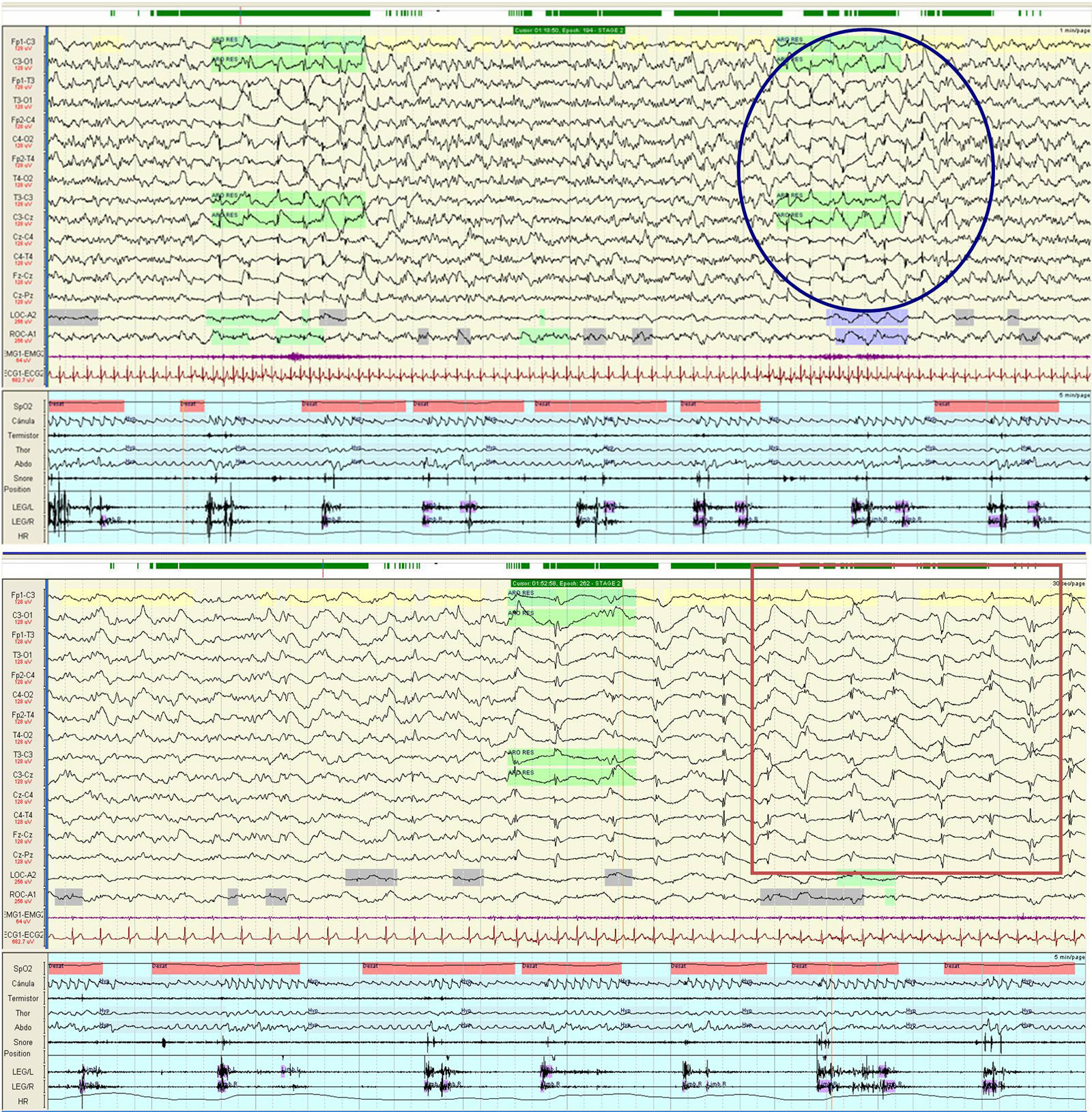
We present the case of a 53-year old man with a personal history of obstructive sleep apnoea-hypopnoea syndrome and a family history of CJD (the patient's father had died of CJD 11 years earlier). He was admitted due to a 10-week history of progressive difficulty speaking, as well as gait instability and abnormal movements of the left arm. His examination revealed language impairment, generalised spasticity, dystonic movements of the left hand, generalised myoclonus, gait ataxia, and pyramidal signs. A brain magnetic resonance imaging (MRI) scan showed abnormal diffusion restriction and increased signal intensity in the bilateral striatum, insular cortex, cingulate cortex, and frontal areas of both hemispheres, predominantly in the right. We performed 2 EEGs, on days 2 and 7 after admission, observing epileptiform activity consisting of 1-Hz spike-and-wave complexes in the right parietal region and at the midline, accompanied on occasions by facial myoclonus and clonic jerks of the left hand, maintaining a reactive posterior alpha rhythm (Fig. 1). The V-PSG study performed one week after admission revealed generalised periodic activity with bi- and triphasic waves at 1–2Hz during wakefulness and arousals, and absence of the EEG readings typical of the REM and non-REM sleep stages, which were characterised by a cyclic alternating pattern (CAP). Furthermore, despite the correct use of continuous positive airway pressure during the reading, the apnoea-hypopnoea index score was higher than 50events/h (Fig. 2). The patient remained hospitalised in the neurology department, with his condition progressing to akinetic mutism, and died 2 months after admission. The genetic study detected the p.Glu200Lys mutation (p-E200K), at position 598 of exon 2 of the PRNP gene, confirming diagnosis of FCJD.
 Frequent epileptiform discharges consisting of 1-Hz spike-and-wave complexes are observed on the right parietal region and midline (C4-P4, P4-O2, Fz-Cz, Cz-Pz), occasionally radiating to the adjacent areas. (B) A parieto-occipital alpha rhythm is recorded in the posterior regions (P3-O1, T5-O1, P4-O2, T6-O2); this rhythm was reactive to the closing of the eyes and attenuated when eyes opened.")
Baseline electroencephalography. (A) Frequent epileptiform discharges consisting of 1-Hz spike-and-wave complexes are observed on the right parietal region and midline (C4-P4, P4-O2, Fz-Cz, Cz-Pz), occasionally radiating to the adjacent areas. (B) A parieto-occipital alpha rhythm is recorded in the posterior regions (P3-O1, T5-O1, P4-O2, T6-O2); this rhythm was reactive to the closing of the eyes and attenuated when eyes opened.
, corresponding to an arousal after a respiratory event. Generalised activity of 1-Hz bi- or triphasic waves was accompanied by increased heart rate. The square indicates phase 2 of the CAP, characterised by diffuse slow waves and lack of physiological signs typical of the different sleep stages.")
Night-time polysomnography. The circle indicates phase 1 of the cyclic alternating pattern (CAP), corresponding to an arousal after a respiratory event. Generalised activity of 1-Hz bi- or triphasic waves was accompanied by increased heart rate. The square indicates phase 2 of the CAP, characterised by diffuse slow waves and lack of physiological signs typical of the different sleep stages.
Our EEG findings are consistent with those published in the literature, according to which the reactive posterior rhythm may still be continued in early stages of CJD. FIRDA and PLED may be the predecessors of PSWC.8 Appel et al.9 found a correlation between periodic activity and cortical involvement in MRI scans of patients with the E200K mutation. Other studies10 report that the incidence of seizures is lower in patients with the E200K mutation than in those with sporadic CJD, due to the involvement of less epileptogenic regions of the deep grey matter.9 As in previous studies, the V-PSG revealed a CAP.11 Cohen et al.1 report severe sleep impairment in patients with the E200K mutation; these findings manifested early during the course of the disease, with the V-PSG study revealing disappearance of sleep spindles, very low sleep efficiency, and lack of REM sleep. A recent study of 28 consecutive patients with post-mortem diagnoses of CJD showed that 90% of the patients reported sleep disturbances at the baseline examination.7 The severe disruption of sleep architecture and the disappearance of sleep spindles have been associated with thalamic involvement.1 Like other researchers, we believe further studies are needed to assess the role of sleep in the early diagnosis of patients with suspected CJD, and especially familial forms.
Please cite this article as: Garnés Sánchez CM, López Bernabé R, Miró-Andreu A, Maeztu Sardiña MC, Salmerón-Ato P. Hallazgos electroencefalográficos y polisomnográficos en un paciente con enfermedad de Creutzfeldt-Jakob familiar. Neurología. 2018;33:625–628.
