



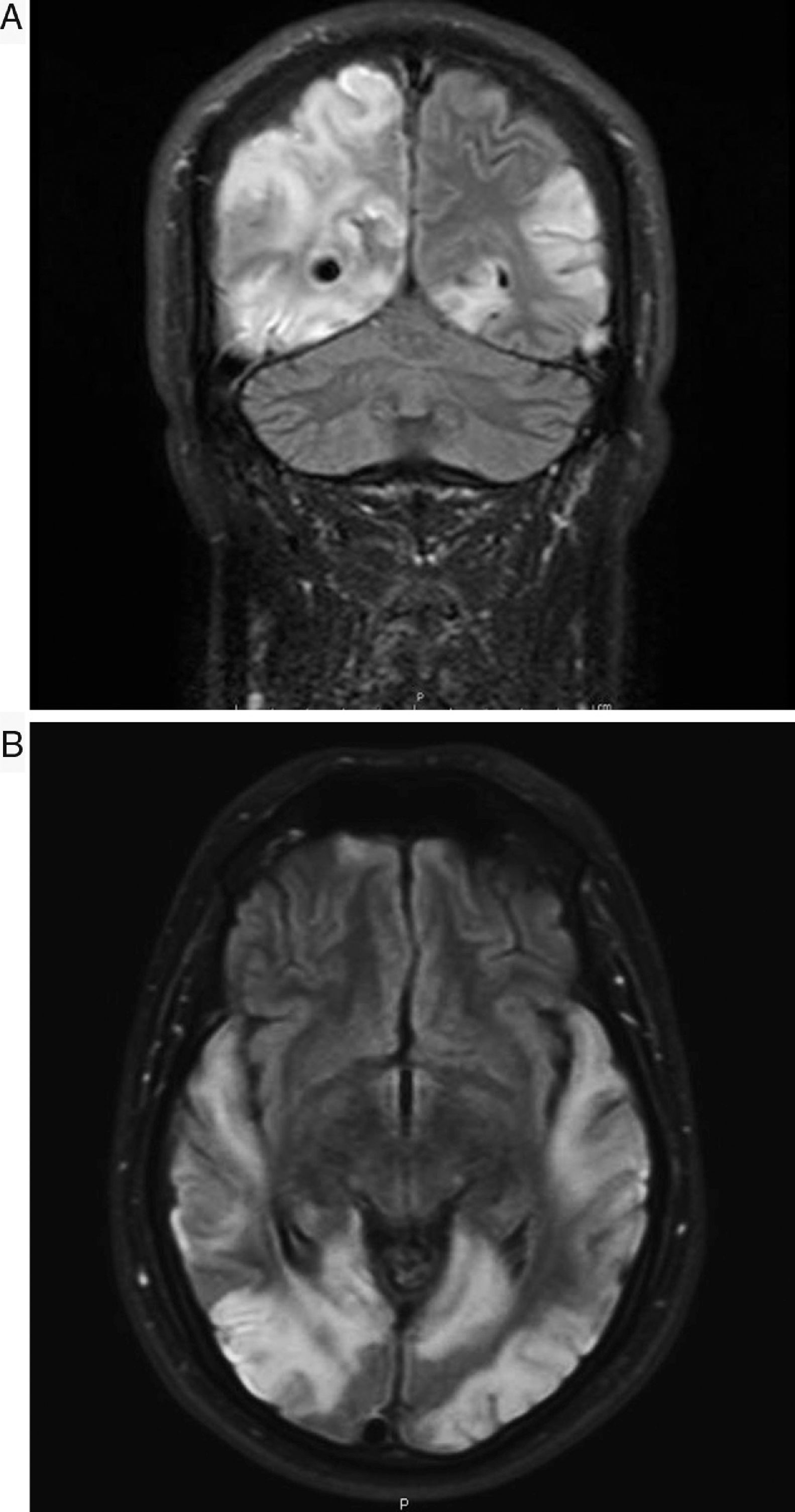
Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) syndrome is classified within the group of mitochondrial diseases. The lack of large series of patients with the same molecular defect and clinical manifestations is a fundamental problem, as these may lead to conclusive studies on the effectiveness of the different drugs applied.1 We present the case of a 30-year-old man with no relevant history who in January 2013 presented an episode compatible with secondarily generalised focal-onset seizures with subsequent status epilepticus, requiring admission to the intensive care unit. A brain MRI scan only revealed focal cortical thickening of the left lingual gyrus, suggesting focal cortical dysplasia as the first diagnostic hypothesis. The patient remained asymptomatic until early September 2014, when he was admitted to our centre due to sudden-onset visual loss affecting the left hemifield. An initial neurological examination only revealed left homonymous hemianopsia and slight dysmetria during the finger-to-nose test with the left arm. The patient presented a surprising, rapid decline during the following days, developing cortical blindness, cortical deafness, and global aphasia. He also presented significant psychomotor agitation and occasional marked agitation with continuous jargon aphasia and unmotivated actions. His only contact with his environment was by touch, and he was able to recognise his wife by touching a ring she was wearing. A laboratory analysis showed high lactate concentrations in the blood (2.7mmol/L) and the cerebrospinal fluid (3.36mmol/L), as well as high blood l-carnitine and total carnitine levels; creatine kinase level was normal. No signs of myopathy were observed in the electromyography study, and a hearing test revealed bilateral mild sensorineural hearing loss. A further MRI scan showed significant lesion extension with cortical involvement in the left hemisphere (predominantly occipital, parietal, and temporal), with hyperintensities on T2-weighted and FLAIR sequences. Oedema, mass effect areas, and diffusion restriction were also observed. The patient also presented occipito-parieto-temporal involvement with similar characteristics in the right hemisphere, and some areas of cortical laminar necrosis (Fig. 1A and B). Muscle biopsy showed non-specific changes and a lack of morphological changes diagnostic of mitochondrial myopathy (no ragged red fibres and no COX-negative fibres). Results of the genetic study were positive for the A3243G mutation. During hospitalisation, the patient was treated with phenytoin 100mg (dosed at 100-50-100mg), levetiracetam 1000mg/12h, clonazepam 0.5mg/12h, ubiquinol 200mg/8h, idebenone 90mg/8h, arginine 6g/8h, and vitamin complexes (thiamine [B1] 300mg [1/2 tablet/12h], riboflavin [B2] 50mg/8h, vitamin C 2g/day; vitamin E 200mg/day on Mondays, Wednesdays, and Fridays). His condition improved to a certain extent. This case presents several interesting features. Firstly, late onset is noteworthy, occurring in approximately 20% of cases. It is also surprising that the patient did not show muscle involvement, which is especially frequent in carriers of the A3243G mutation. Various studies have reported that the A3243G mutation provokes a severe combined defect in the respiratory chain of myoblasts. Muscle biopsies normally reveal fat deposits, and ragged red fibres are also observed frequently. According to some studies, the percentage of patients diagnosed with MELAS syndrome (regardless of the mutation) after a negative biopsy is approximately 10%.2 The aggressive symptom progression is also of interest, with the patient progressing from hemianopsia to blindness, deafness, and mixed aphasia in only 3 days, which made him practically unable to communicate with his environment. Central deafness has very rarely been reported.3 Regarding treatment, vitamin complexes (B1, B2, C, and E) would act as antioxidants in this case, which appears to correct the oxidative damage; however, there are no conclusive studies on the effectiveness of this treatment.4 We should also stress the administration of l-arginine, a nitric oxide precursor; several studies report a decrease in its levels both in the acute phase and in the interictal phase, compared to controls. Arginine is considered one of the most promising drugs5,6 and, although its action mechanisms are not fully understood, it seems to have an impact on vascular regulation, causing vasodilation by increasing nitric oxide concentration. This would increase aerobic capacity and improve muscle metabolism.6 Arginine can be administered by intravenous infusion in the acute phase and acts rapidly (less than 24h), improving symptoms. However, symptoms may worsen subsequently if oral supplementation is not continued, with a recommended dose of 0.5g/kg/day. Our patient is currently receiving oral arginine at 6g/8h. The patient was hospitalised for approximately one month and a half, and showed some degree of improvement. At discharge, he was able to follow simple instructions and produce short, coherent sentences. His vision improved slightly, and he is currently able to identify colours, shapes, and movement. Speech impairment with paraphasia persisted and the patient continued receiving speech therapy on an outpatient basis. No motor deficit was observed at any time. He continued treatment with vitamin complexes, oral arginine 6g/8h, clonazepam 0.5mg/8h, levetiracetam 500mg/12h, and lacosamide 100mg/8h; phenytoin was suspended. The patient remains stable and has not been readmitted again to date.
 and axial (B) FLAIR sequences showing hyperintense lesions affecting the bilateral temporo-parieto-occipital cortex, which do not correspond to vascular territories.")
Please cite this article as: Pérez Torre P, Acebrón-Herrera F, García Barragán N, Corral Corral Í. Afectación encefálica global y uso de L-arginina en un paciente con síndrome de MELAS. Neurología. 2020;35:435–437.
