



Amyotrophic lateral sclerosis (ALS) is a disease with very poor prognosis, and a mortality of 50% at 18 months after diagnosis. Multidisciplinary units attempt to improve the quality of life and survival of patients with ALS. The aim of this study is to evaluate every 3 months, over a 24-month period, the outcome of patients treated at the ALS unit since the time of diagnosis.
Material and methodsWe performed a prospective observational study of patients treated in the ALS unit following a clinical pathway since the time of diagnosis with quarterly reviews from 2006 to 2010. The age of onset, functional impairment (ALSFRS-r), impairment of respiratory function, dysphagia and signs of depression and/or cognitive impairment were evaluated in relation to the initial location symptoms (bulbar [B], upper limbs [UL], lower limbs [LL]).
ResultsA total of 42 patients (30 males and 12 females) were evaluated (mean age at onset of 57.97 years old, SD 14.56). There was an even distribution by location of onset of symptoms (B 14 patients, UL 14, LL 14.) Functional impairment (B −26.89 points, UL −22.48 points, LL −22.66 points), the need for use of BIPAP (B 64.28%; UL 35.71%; LL 50%), the presence of dysphagia (B 85.71; UL 42.85; LL 71.42%), signs of depression (B 78.57%; UL 35.71%; LL 64.28%) and cognitive impairment (B 42.85%; UL 21.42; LL 35.71%) were higher at 24 months of progression in patients with bulbar onset. There was no difference in mortality data (23.80% overall).
ConclusionsThe treatment in multidisciplinary units does not change the neurological progression of the disease, but increases the survival of ALS patients regardless of their initial onset, emphasising the use of multidisciplinary care.
La esclerosis lateral amiotrófica (ELA) es una enfermedad con muy mal pronóstico, con una mortalidad del 50% a los 18 meses tras el diagnóstico. Las unidades multidisciplinares pretenden mejorar la calidad de vida y la supervivencia de los enfermos de ELA. El objetivo de nuestro estudio es evaluar cada 3 meses la evolución de pacientes atendidos en la unidad de ELA desde el momento del diagnóstico y durante 24 meses.
Material y métodosSe realizó un estudio observacional prospectivo de pacientes atendidos en la unidad de ELA siguiendo una vía clínica desde el momento del diagnóstico y con revisiones trimestrales desde 2006 a 2010. La edad de inicio, el deterioro de la situación funcional (escala ALSFRS-r), el deterioro de la función respiratoria y la aparición de disfagia y de signos de depresión y/o de deterioro cognitivo fueron evaluados en relación con la localización inicial de los síntomas (bulbar [B], miembros superiores [MMSS], miembros inferiores [MMII]).
Resultados42 pacientes (30V y 12M) fueron evaluados (edad media de inicio±desviación estándar de 57,97±14,56 años). Se encontró una distribución igual por localización de inicio de los síntomas (B 14 pacientes, MMSS 14, MMII 14). El deterioro funcional (B –26,89 points; MMSS –22,48 points; MMII –22,66 points), la necesidad de uso de BIPAP (B 64,28%; MMSS 35,71%, MMII 50%), la presencia de disfagia (B 85,71; MMSS 42.85; MMII 71.42%), de signos de depresión (B 78,57%, MMSS 35,71%; MMII 64,28%) y de deterioro cognitivo (B 42,85%; MMSS 21,42; MMII 35,71%) fue mayor a los 24 meses de evolución en los pacientes de inicio bulbar. No hubo diferencias en los datos de mortalidad (global 23,80%).
ConclusionesEl tratamiento en unidades multidisciplinares no varía la evolución neurológica de la enfermedad pero favorece la aplicación de cuidados multidisciplinares e incrementa la supervivencia de los enfermos de ELA independientemente de su forma de inicio.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by signs and symptoms of primary degeneration of superior and inferior motor neurons. Over the course of the disease, progressive weakness and wasting occur in the muscles innervating the bulbar, chest, abdomen and limbs.
The risk of developing ALS over one's lifetime is 1:1000. The condition's incidence is estimated at between 1.5 and 2.7 for every 100,000inhabitants/year in Europe and North America, with a slight predominance in males (1.5:1) and a mean age of 64 years at onset of symptoms. Death is usually due to respiratory failure occurring between 3 and 5 years after the start of symptoms; according to historical series, 50% of patients die within the 18 months following diagnosis.1–4
There is currently no curative treatment for ALS and riluzol is the only specific therapy to increase survival slightly. Patient management is essentially based on palliative treatment and symptom control,5–7 including the use of medication and the implementation of percutaneous gastrostomies and invasive or non-invasive ventilation systems.
In the last few years, multidisciplinary units have emerged around the world7,8 for the treatment of these patients. The accumulation of a large number of patients leads to the grouping of resources and clinical experts to facilitate their treatment. Although these multidisciplinary units improve quality of life and extend survival in other neurodegenerative diseases, their effect on ALS is not yet clear.7–11
A network of multidisciplinary units specializing in the diagnosis, management and palliative care of ALS was set up in Madrid (Spain) in 2006. Patient care was protocolized through the development of a clinical pathway that included aspects of diagnosis, treatment and care, from the viewpoints of both medicine and social attention.12
The aim of the present study is to analyze the data on ALS patients treated at one of the units in the network under the clinical pathway protocol applied every 3 months during the first 2 years following diagnosis.
Material and methodsWe have carried out a descriptive observational study of patients seen at the ALS multidisciplinary unit between March 2006, and January 2010.
Those selected for further study were the patients diagnosed with definitive or probable ALS, according to the revised El Escorial criteria,13 who regularly attended, at least every 3–4 months, for a follow-up visit at the multidisciplinary unit. Those patients who did not complete at least 1 year of follow-up after diagnosis were excluded. The maximum monitoring period in the study was 2 years.
All patients selected were treated with riluzol throughout the follow-up period, although this was not an inclusion/exclusion factor under study. At the multidisciplinary unit, following the pre-established clinical pathway,12 they received neurological, psychological, palliative and social care; respiratory and nutritional studies were performed, with gastrostomies or non-invasive ventilation systems being adopted and motor, respiratory and phoniatric rehabilitation being provided in line with the patients’ individual needs from the moment of diagnosis.
The data analyzed included age and the initial location of the symptoms, the functional impairment observed each quarter (using the ALSFRS-r scale14), the deterioration in their respiratory function (FVC<70%), the appearance of dysphagia (more than 2 on the Karnell scale15) and signs of depression (more than 21 points on the BDI scale16) and/or cognitive impairment (Mini Mental State Examination <24/30) during the first 2 years following diagnosis.
The institution of non-invasive ventilation systems and gastrostomies was not determined by any single test but rather by the combination of different variables that may vary in each patient (fall in FVC, night-time pulsioxymetry desaturation index, PIM and PEM values for the ventilation systems, rapid weight loss, intense dysphagia, dehydration in the case of gastrostomies).
These data were assessed in connection with the initial location of the symptoms as follows: bulbar (B), upper limbs (UL) or lower limbs (LL).
The statistical study was performed using the PASW Statistics 18 computer application for statistics, using the χ2 test for the differences between categorical variables and the McNemar test for paired variables, and generating a mean least squares analytical model. Values were considered significant when p<0.05.
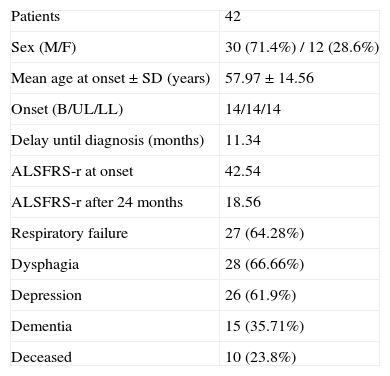
ResultsForty-two patients with a definitive or probable diagnosis of ALS were selected following the criteria set out above from among the 97 people seen at the ALS unit between March 2006, and January 2010. Of these, 30 were males and 12 females, the mean age of onset of the symptoms±standard deviation was 57.97±4.56 years and the delay until diagnosis can be estimated at 11.34 months (Table 1).
Distribution of the sample.
| Patients | 42 |
| Sex (M/F) | 30 (71.4%) / 12 (28.6%) |
| Mean age at onset±SD (years) | 57.97±14.56 |
| Onset (B/UL/LL) | 14/14/14 |
| Delay until diagnosis (months) | 11.34 |
| ALSFRS-r at onset | 42.54 |
| ALSFRS-r after 24 months | 18.56 |
| Respiratory failure | 27 (64.28%) |
| Dysphagia | 28 (66.66%) |
| Depression | 26 (61.9%) |
| Dementia | 15 (35.71%) |
| Deceased | 10 (23.8%) |
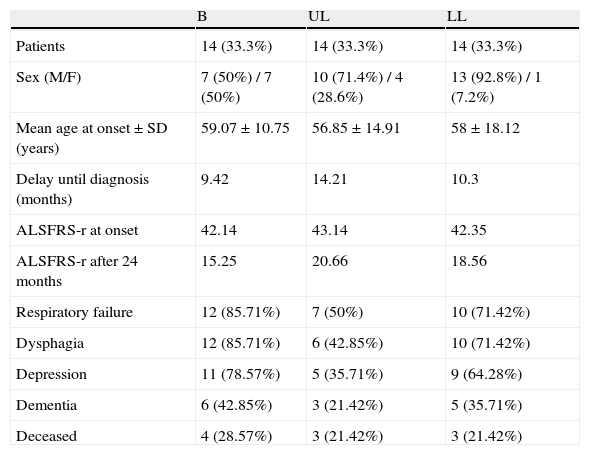
A similar distribution was found in the 3 groups created according to the initial location of the debut symptoms (14 patients in each group). The patients in the B group were 7 males (50%) and 7 females (50%), with a mean age of onset of the symptoms was 59.07 years (M 55.57 versus F 62.57). Those in the UL group were 10 males (71.42%) and 4 females (28.57%), with a mean age of onset of 56.85 years (M 57.10 versus F 56.25). Finally, those in the LL group were 13 males (92.85%) and 1 female (7.14%), and the age of onset was 58.74 years (M 57.92 versus F 59). The age differences between the groups were not statistically significant (p>0.05). The delay in diagnosis was less in patients with bulbar onset, but with significant differences (Table 2).
Distribution of the sample by groups.
| B | UL | LL | |
| Patients | 14 (33.3%) | 14 (33.3%) | 14 (33.3%) |
| Sex (M/F) | 7 (50%) / 7 (50%) | 10 (71.4%) / 4 (28.6%) | 13 (92.8%) / 1 (7.2%) |
| Mean age at onset±SD (years) | 59.07±10.75 | 56.85±14.91 | 58±18.12 |
| Delay until diagnosis (months) | 9.42 | 14.21 | 10.3 |
| ALSFRS-r at onset | 42.14 | 43.14 | 42.35 |
| ALSFRS-r after 24 months | 15.25 | 20.66 | 18.56 |
| Respiratory failure | 12 (85.71%) | 7 (50%) | 10 (71.42%) |
| Dysphagia | 12 (85.71%) | 6 (42.85%) | 10 (71.42%) |
| Depression | 11 (78.57%) | 5 (35.71%) | 9 (64.28%) |
| Dementia | 6 (42.85%) | 3 (21.42%) | 5 (35.71%) |
| Deceased | 4 (28.57%) | 3 (21.42%) | 3 (21.42%) |
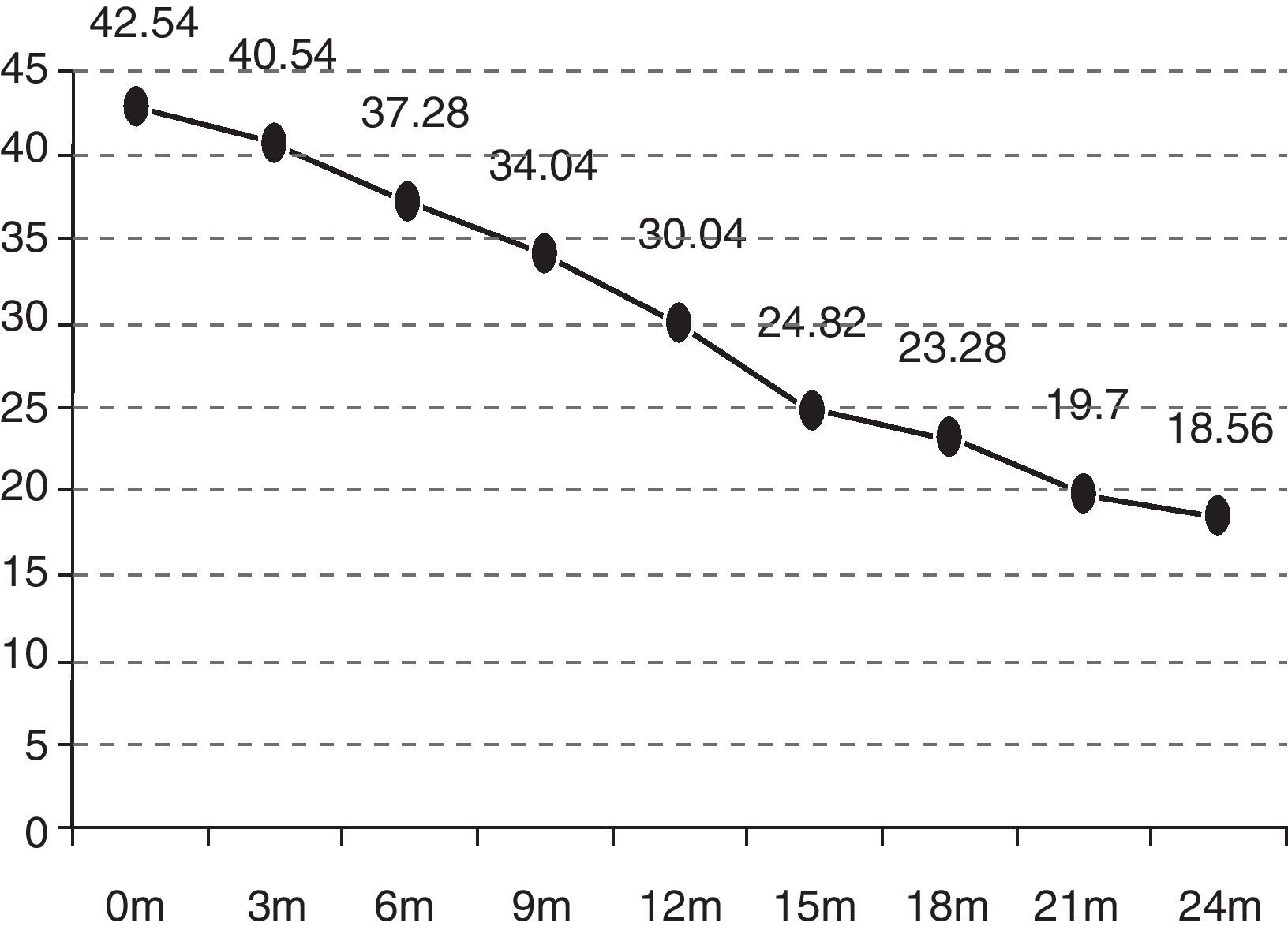
In the quarterly follow-up of patients, it was possible to observe how the functional decline is statistically significant after 6 months of follow-up (p=0.0297). The initial functional status (ALSFRS-r scale) of the patients was 42.54 points and deteriorated to 18.07 after 24 months (−24.47 points) (Fig. 1).

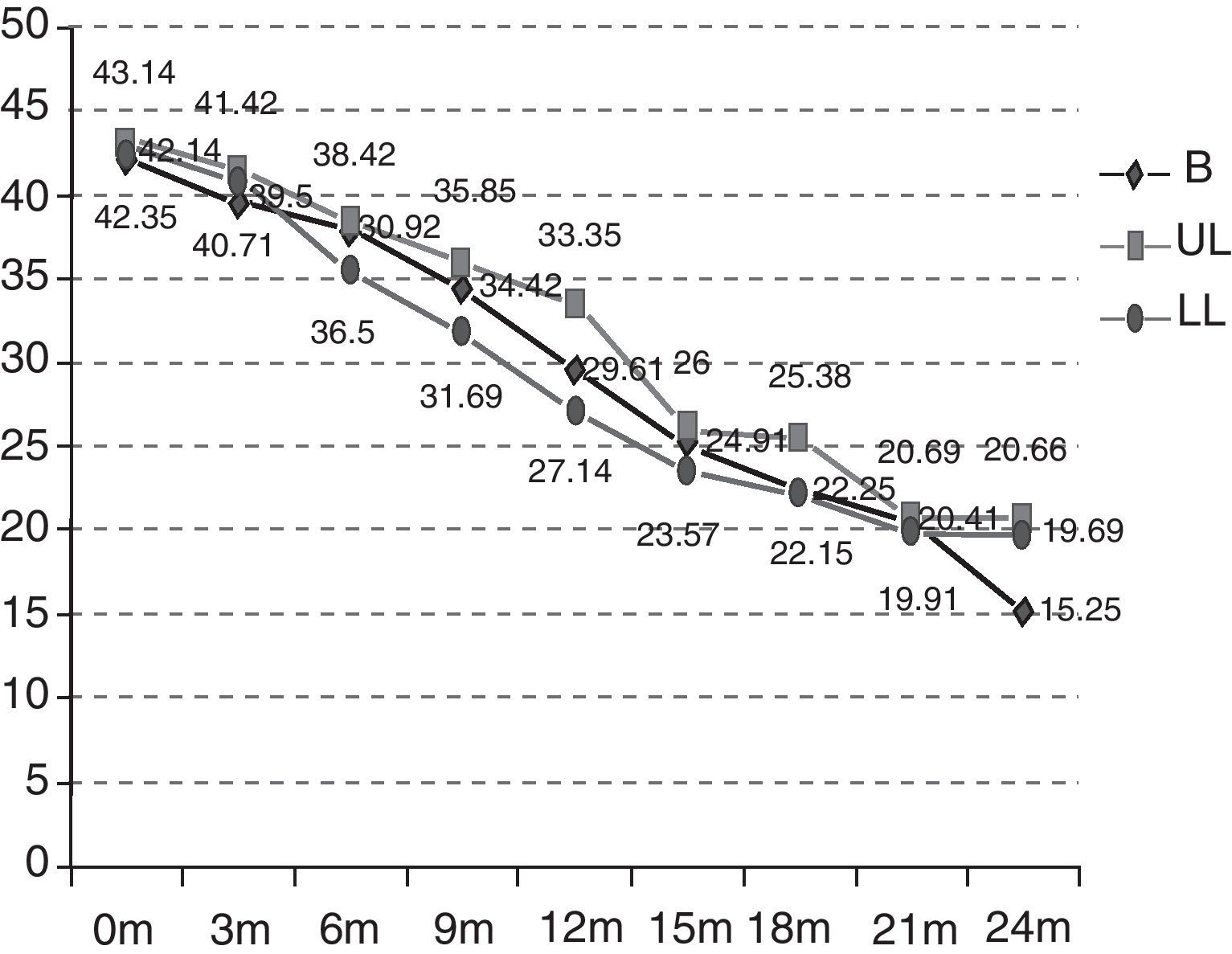
As can be observed in Fig. 2, the initial functional status of the patients in the B group was 42.14 ALSFRS-r points on average versus 43.14 and 42.35 points for those in the UL and LL groups. After 24 months, the functional status was also worse in the rest of the groups (B 15.25 versus UL 20.66 versus LL 19.69) and, therefore, the mean decrease over these 24 months was also more marked (B −26.89 versus UL −22.48 versus LL −22.66) (p>0.5). The deterioration is significant in the different groups after 12 months (the B group p=0.0059; UL p=0.0197; LL p=0.0003). The differences between the groups studied are not statistically significant (p=0.9971).

Respiratory failure symptoms affected 69.04% of patients; the establishment of non-invasive mechanical ventilation was prescribed in 54.76% of all patients, 79.3% of whom had a FVC of less than 70%.
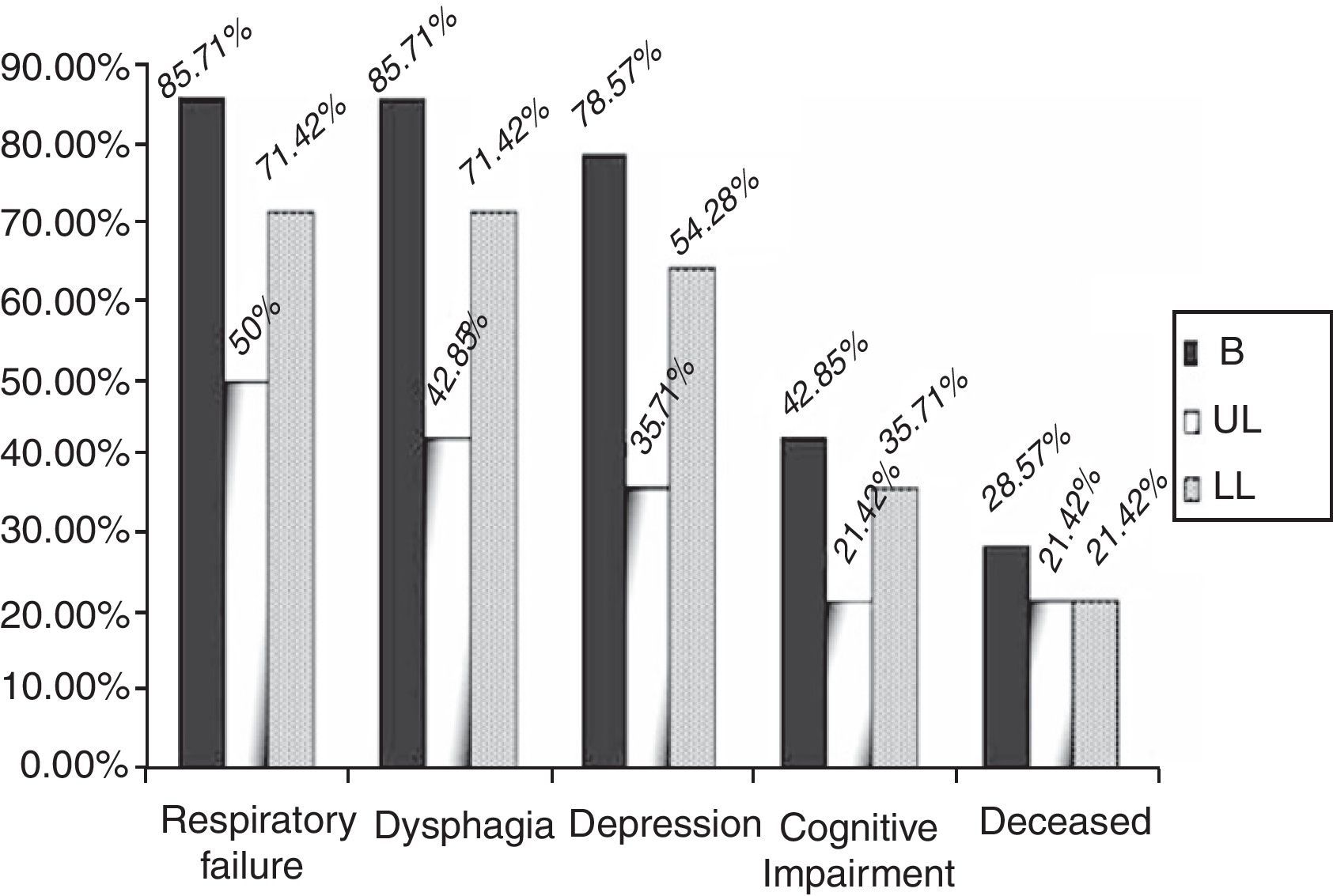
By groups, 12 (85.71%) of the patients in the B group suffered a significant decrease in respiratory function, versus 7 (50%) (p=0.04) of the patients in the UL group and 10 (71.42%) (p=0.43) in the LL group over the 2 years of follow-up (Fig. 3). In addition, the start of treatment with a non-invasive mechanical ventilation device was prescribed, although they ultimately may not have used it, for 9 (64.28%) of the patients in the B group, 5 (35.71%) in the UL group and 7 (50%) in the LL group. These data are not globally significant (p=0.309).

Dysphagia was present in 28 patients (66.6%), 12 of the patients in the B group (85.71%) versus 42.85% (6 patients) in the UL group and 71.42% (10 patients) in the LL group (Fig. 3) (p=0.355). A gastrostomy was applied to 46.42% of the patients with dysphagia (30.95% of the overall total).
With respect to symptoms of depression or cognitive alterations, these were more noticeable in the B group patients. Of the patients in the B group, 78.57% (11) presented depressive symptoms reactively from the start of the illness. Five of these 11 patients also had signs of cognitive impairment and there was also one case without any associated symptoms of depression. The 6 patients with signs of cognitive impairment represented 42.85%.
In the UL group, 5 patients (35.71%) (p=0.250) had depression symptoms, associated in 3 cases with signs of cognitive impairment (21.42%); in the LL group, there were 9 (64.28%) (p=0.50) with depression symptoms and 5 (35.71%) had signs of cognitive impairment (Fig. 3) (p=0.87).
There were no great differences in the mortality data, 4 patients (28.57%) died in the B group, 3 (21.42%) in the UL group and 3 (21.42%) in the LL group over the 2 years of the study (p>0.05), which represents a global mortality of 23.80%.
DiscussionThe data from before the ALS unit was set up are rather incomplete and biased. Patients were often not followed up after diagnosis and there are no data on the progress of their neurological or pneumological functional status or any neuropsychological evaluation. Resorting to the pharmacy records at our hospital, we were able to analyze in due course the reports of patients who had been taking riluzol in the years prior to the opening of the unit.17 These data show how the delay in diagnosis has come down from 27.61 to 11.34 months since the onset of symptoms and that the implementation of ventilation systems (11.5% pre-unit) and gastrostomies (8.6% pre-unit) and the percentage of patients seen for depression symptoms (10.6% pre-unit) were all much lower than shown by the data currently obtained. No reliable mortality data are available for that pre-unit period.
Although it was not the purpose of this study, the most striking aspect of the patient records in the ALS unit is the mortality figures. The number of deaths after 24 months of follow-up is clearly lower than is shown in articles reviewing the clinical course of the disease, where mortality is calculated to be 50% after 18 months1–4 and lower than the data found in various papers designed to assess whether multidisciplinary units were useful for improving patient survival.9,11,18
Thus, in the study carried out on the multidisciplinary units in Ireland,18 it was concluded that ALS patients seen at multidisciplinary units versus those treated at general neurology departments died later and those patients whose symptoms had begun at the bulbar level particularly benefited from multidisciplinary care. Despite this, 2-year mortality was around 50% globally and 57% for bulbar cases.
In the study undertaken by Chio et al.9 to assess the impact of care and on-going visits to a third-level reference hospital on the prognosis for the disease, the mean survival of the patients seen at a third-level ALS centre is 1080 days, but there are no figures for the percentage of mortality after 2 years.
The data were worse in the study conducted on multidisciplinary units in Southern Italy,11 with a mortality of 24% in patients over the first 12 months. In this paper, it was striking that over 30% of patients were not under treatment with riluzol.
Therefore, our findings of a 2-year mortality of only 23% of patients, 29% in the case of those with bulbar onset, are clearly better. Several factors may explain this.
Our patients are younger than those in other studies9,11,18 (58 years versus 60. 61 and 64 years of age, respectively). Various papers19–21 have evaluated the natural course of ALS in young patients and have concluded that survival is higher in such cases. However, the patients in our study do not fit in the category of “young” applied in these studies which recruited patients under 2519 or under 4019–21 and the difference in the mean ages with respect to the studies for other multidisciplinary units9,11,15 is very small. On the other hand, the patients in these prior studies, apart from being younger, had a predominance of superior motor neuron symptoms or a considerable interval between the onset of symptoms and their diagnosis, both matters that are not true in our study.
Perhaps the fundamental factor we should consider is the strict monitoring of patients as per the protocol applied in our setting.12 The Irish network of units18 also has intensive monitoring of patients, but every 6 months and, on occasions, by telephone, so it is not possible to guarantee the level of care given in some aspects of the illness. On the other hand, it is the only one to have a polished network of social and palliative care like ours. The Italian studies9,11 speak about care provision at units but do not have a protocol with standard follow-up intervals.
These data would be in line with the recommendations of the AAN and the EFNS22,23 about the potential of protocols to optimize patient care and improve their survival. In this way, the establishment of ventilation systems for patients with respiratory failure and gastrostomies was clearly higher than that reflected in the first records24 following the publication of the AAN's clinical guidelines and might explain the greater survival of our patients.
At the neurological functional level, it is not possible to speak of differences between groups although there is a slight tendency towards a greater decrease in the score on the ALSFRS-r scale in the B group. The impairment curves are not very different from those recently observed by Gordon in ALS patients not monitored at multidisciplinary units.25
With respect to the rest, the results of multidisciplinary involvement show a trend towards a worse respiratory prognosis and a greater need for care among patients with clinical signs of bulbar onset compared to patients with spinal onset, although the small size of the sample does not allow statistically significant differences to be obtained. It is striking that the results obtained in patients with initial involvement of the lower limbs are worse than those with initial onset in the upper limbs, although the differences are small.
The worse prognosis for patients with bulbar involvement has been shown and discussed in several papers26–28 that have revealed the existence of confounding factors, such as the age of onset of symptoms, as these patients are normally older. In our case, there was no clear age difference between the groups and, therefore, we considered the trend towards a worse evolution in patients with bulbar clinical involvement compared to those with spinal predominance to be very notable. However, we could not say there was any increase in mortality in those patients with respect to spinal ones during the first 2 years.
We believe that the high levels of patients with symptoms of depression and cognitive impairment are due to the fact that the patients are monitored at a multidisciplinary unit12 and their follow-up by different specialists according to the protocol might make it easier to detect sub-clinical findings of these phenomena more often than in other papers.29,30
In conclusion, we can state that treatment at multidisciplinary units does not seem to alter the neurological course of the disease but rather it favours the application of respiratory and nutritional care, as well as the detection and treatment of symptoms of depression and cognitive impairment. Thanks to all of these factors, survival among ALS patients treated at a multidisciplinary unit is greater, regardless of the type of onset.
Conflicts of interestThe authors have no conflict of interests to declare.
Please cite this article as: Rodríguez de Rivera FJ, et al. Evolución de pacientes con esclerosis lateral amiotrófica atendidos una unidad multidisciplinar. Neurología. 2011;26:455–60.
