



The variant c.1414-1G>T in the GRN gene has previously been reported as probably pathogenic in subjects of Hispanic origin in the American continent.
MethodsWe report 5 families of Spanish origin carrying this variant, including the clinical, neuroimaging, and laboratory findings.
ResultsPhenotypes were strikingly different, including cases presenting with behavioral variant frontotemporal dementia, semantic variant primary progressive aphasia, rapidly progressive motor neuron disease (pathologically documented), and tremor-dominant parkinsonism. Retinal degeneration has been found in homozygous carriers only. Ex vivo splicing assays confirmed that the mutation c.1414-1G>T affects the splicing of the exon, causing a loss of 20 amino acids in exon 11.
ConclusionsWe conclude that variant c.1414-1G>T of the GRN gene is pathogenic, can lead to a variety of clinical presentations and to gene dosage effect, and probably has a Spanish founder effect.
La variante c.1414-1G > T en el gen GRN ha sido reportada previamente como probablemente patogénica en sujetos del continente Americano de origen Hispano.
MétodosReportamos cinco familias de origen Español portadoras de la mencionada variante. Se presentan las características clínicas, de neuroimagen y de laboratorio.
ResultadosLos fenotipos encontrados difieren llamativamente entre los distintos casos, incluyendo presentación como demencia frontotemporal variante conductual, variante semántica de afasia progresiva primaria, enfermedad de neurona motora rápidamente progresiva (con confirmación neuropatológico) y parkinsonismo de predominio tremórico. Degeneración retiniana fue evidenciada únicamente en portadores homocigotos. Ensayos de splicing ex vivo confirman que la mutación c.1414-1G > T afecta el splicing del exón, causando una pérdida de 20 aminoácidos en el exón 11.
ConclusionesConcluimos que la variante c.1414-1G > T del gen GRN es patogénica, puede causar presentaciones clínicas diversas, efecto de dosis génica y probablemente represente un efecto fundador de origen Español.
Frontotemporal lobar degeneration (FTLD) is characterized by remarkable heterogeneity from a clinical, pathological, and genetic point of view. Progranulin (GRN), microtubule-associated protein tau (MAPT), and chromosome 9 open reading frame 72 (C9orf72) account for most familial cases.1,2GRN mutations account for 4.5% to 10% of FTLD cases, but this proportion increases to 22% in patients with positive family history.3GRN mutations may cause any subtype of FTD, including behavioral variant (BvFTD) and different forms of primary progressive aphasias (PPA).4,5 Patients often present with an extensive phenotypic variability, even among different members of the same kindred carrying an identical disease mutation.6–10
The GRN gene encodes a secreted precursor protein (progranulin, PGRN) composed of a signal peptide and 7.5 tandem repeats of highly conserved motifs containing 12 cysteines that can be proteolytically cleaved to form a family of granulin peptides A to G. Their biological roles, mediated by either the whole protein or its proteolytic cleavage product, include cell proliferation, inflammation, lysosome function, neurite outgrowth, neuronal survival, and metabolic homeostasis.11
Most GRN mutations identified to date are predicted to cause the pathology via a haploinsufficiency mechanism by creating premature stop codons, which in turn result in nonsense-mediated decay (NMD). How a decrease in the levels of GRN causes neurodegenerative processes is not fully understood, as different potential pathways might be involved. PGRN has anti-inflammatory properties, while granulin peptides have pro-inflammatory properties. The upregulation of GRN in activated microglia suggests PGRN might be involved in neurodegeneration through proinflammation.12 Finally, GRN mutant neurons have reduced lysosomal GCase activity, leading to impaired processing of prosaposin, which also may contribute to pathogenesis from GRN mutations.13
To date, more than 170 GRN mutations have been described, most of which led to reduced function or decreased expression of the protein PGRN.14,15 Among these, there has been described an infrequent splice site variant c.1414-1G>T in 2 Hispanic families in the American continent. The variant is predicted to affect an acceptor splice site and is categorized as a likely pathogenic variant.12 Although no studies of progranulin levels in plasma or cDNA have been published, a similar variant (c.1414-2A>G) affects the same core splice-site, and ex vivo splicing assays confirmed that the mutation c.1414-2A>G affects splicing of the exon.16 Moreover, homozygous carriers of this variant (members of one of the families described in this report) were previously reported to develop retinal degeneration, hearing loss, and cognitive impairment in the spectrum of Usher syndrome.19
Our work describes the clinical and neuroimaging features of 5 Spanish families carrying the variant c.1414-1G>T in the GRN gene. We present the clinical, neuroimaging, and laboratory findings, including plasmatic progranulin levels and an RNA study when available. We also present the neuropathologic findings in one case.
MethodSubjectsSubjects were assessed at the Neurology Service of Hospital Universitario Central de Asturias (Oviedo), and referred for genetic analysis to the Genetics Laboratory of Hospital Universitario Central de Asturias. Written informed consent was given for all index cases or their legal guardian according to the Declaration of Helsinki. The informed consent form for genetic studies was approved by the Ethical Committee of Principado de Asturias.
The diagnostic workup of index cases included patient and family medical records, detailed history-taking, neurological examination, cognitive assessment, and neuroimaging as decided by the clinician in charge of the patient. Genealogical trees were drawn from the information provided by index cases or their relatives. Genetic testing was made for the most common genes leading to cognitive impairment, including c9orf72, GRN, MAPT, PS1, PS2, and APP.
Genetic counseling was offered to the relatives of the index cases.
Genetic testingThe genomic DNA was isolated from peripheral blood leukocytes. In the diagnostic procedure of all index cases, NGS (Next Generation Sequencing) for genes associated with neurodegenerative diseases was performed, according to previously published procedures17 (Supplementary file 1).
In silico predictionMaxEntScan was used to predict the splicing effects.18 The variant was classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (https://www.acmg.net/)
Plasma progranulin dosageTo determine the level of plasma progranulin, an enzyme-immunoanalytical essay was performed in an external laboratory (Reference Laboratory, Spain). The normal range level in this laboratory is 21.8-53.2 ng/mL.
GRN mRNA and cDNA analysisRNA from peripheral blood leukocytes was extracted using TRI Reagent™ Solution (Thermo Fisher Scientific, Baltics UAB, Vilnius, Lithuania; AM9738) and was subsequently used for cDNA synthesis with RNA ImProm-II Reverse Transcriptase (Promega Corporation, Madison, WI, USA; A3802), according to the manufacturer’s instructions. To study the effect of the c.1414-1G>T variant on the GRN transcript, we performed a polymerase chain reaction using primers reported in Supplementary material 1. The amplification product was sequenced on an Applied Biosystems ABI3130 Genetic Analyzer using the BigDye Terminator 1.1 Cycle Sequencing Kit according to the manufacturer’s instructions (Thermo Fisher Scientific).
ResultsMolecular findingsTargeted NGS was performed, and we identified 7 symptomatic and 2 asymptomatic heterozygous carriers of the variant c.1414-1G>T in the GRN gene (NM_002087). No other variants were identified. Ex vivo splicing assays showed a loss of 20 amino acids in exon 12 in c.1414-1G>T carriers (p.Val468_Ala492del; c. c1417_1476del) (supplementary, Fig. 1).
Clinical findingsNext, we summarize the main clinical features and neuroimaging findings of the 5 families where we found a variant c.1414-1G>T in the GRN gene (Supplementaryfile 1). All subjects were white, of Spanish origin, were born in the Asturias region (North Spain) ,and were living in Spain or other European countries at the time of diagnosis. In some families, we have been able to draw the full family tree, while in others we report the index case only, with little information on other family members because there were no other members affected or information was inaccessible.
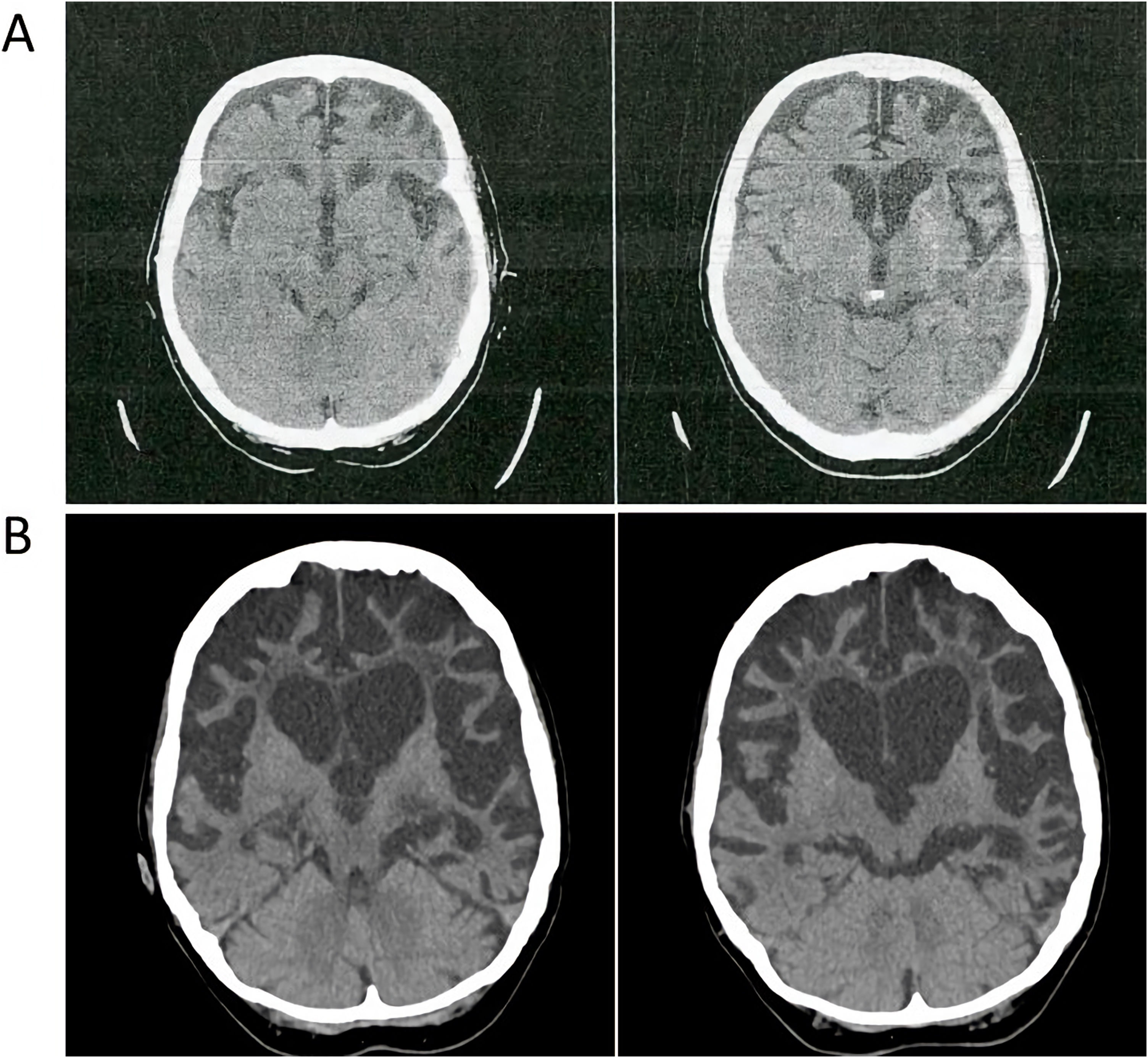
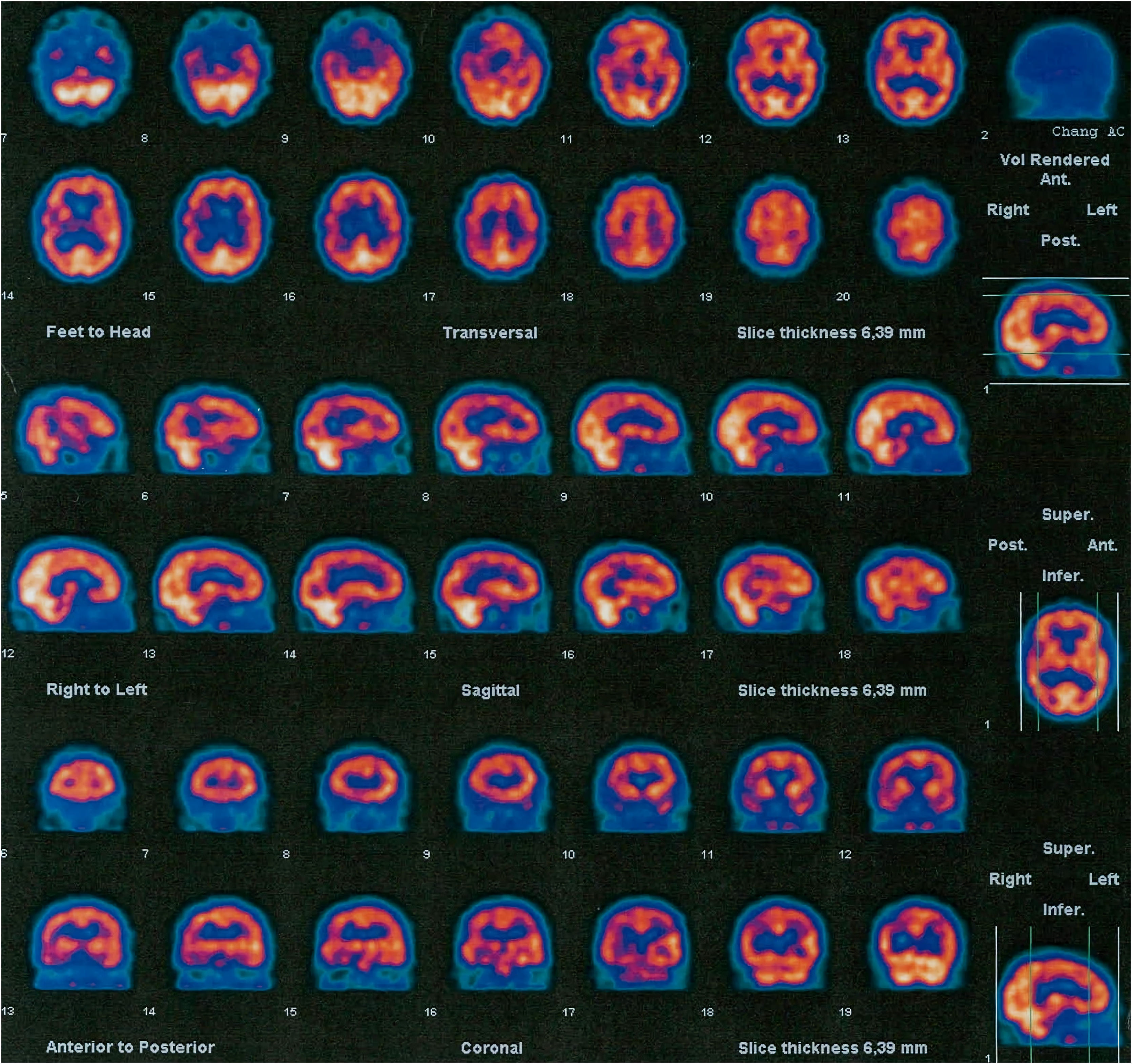
Family #1: The index case is a woman who started at 59 years old with progressive behavioral changes dominated by apathy, perseverative behaviour, hyperorality, lack of self-care, and emotional flattening; and soon later aphasia and dysexecutive syndrome leading to severe dementia in 4 years. A brain CT scan showed bilateral but asymmetric frontotemporal atrophy, much more marked on the left side, that progressed to severe, global, asymmetric atrophy in 5 years (Fig. 1). A brain SPECT scan showed severe hypoperfusion in the frontotemporal lobes. She was diagnosed with behavioral variant FTD (BvFTD) and died 12 years after onset. There were no family members diagnosed with neurodegenerative diseases. An aunt had been diagnosed with a psychiatric disorder in early adulthood, but we were unable to obtain more information on that case.
 Baseline, at diagnosis. B) Follow-up, 5 years after diagnosis.")
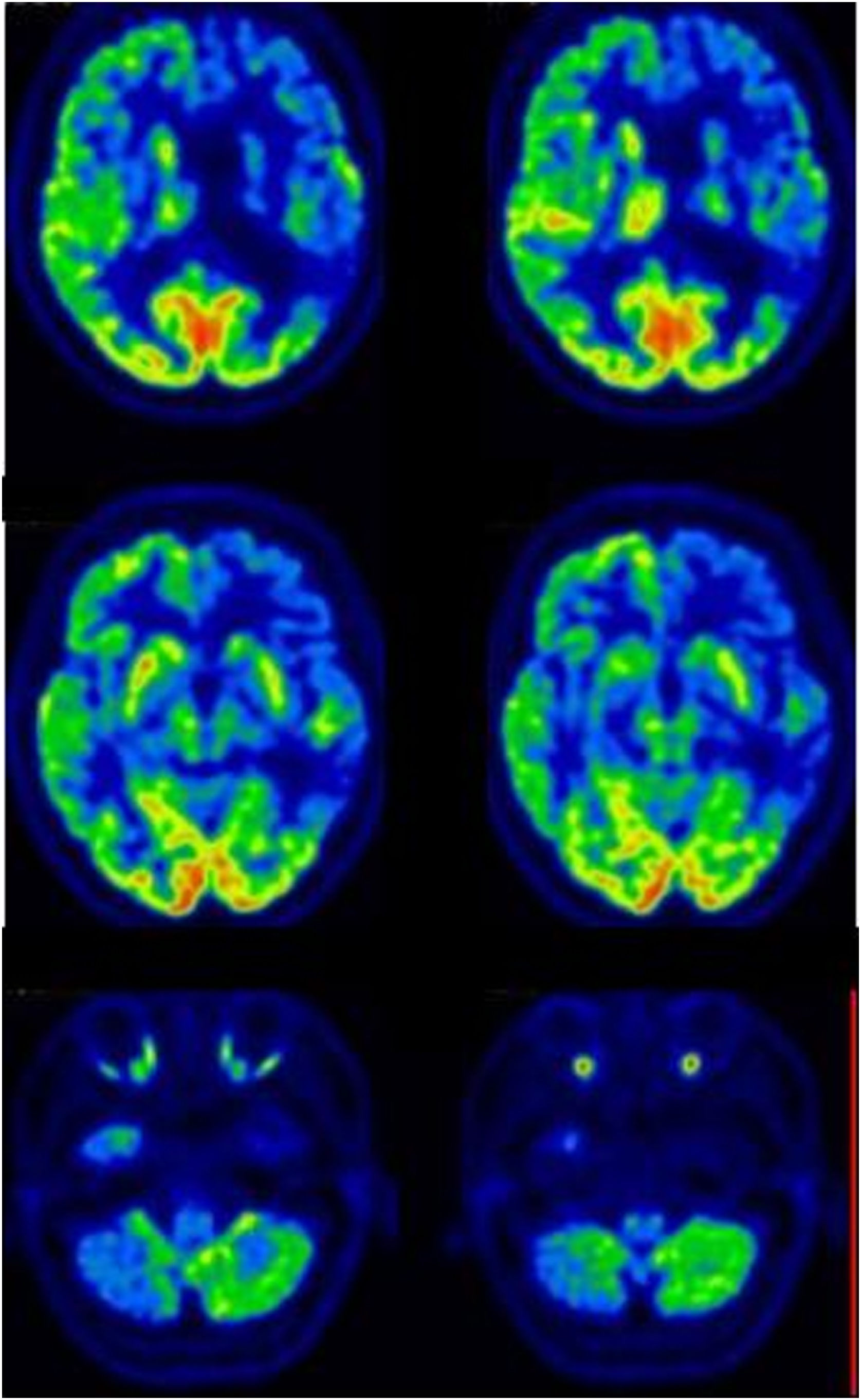
Family #2: The index case is a 69-year-old woman with no relevant family history. She started with difficulties naming objects and comprehending some words. Neuropsychological assessment revealed low scores in naming drawings and surface dyslexia with spared repetition and speech. She was diagnosed with semantic variant primary progressive aphasia (svPPA). The brain CT and MRI scans performed at diagnosis showed asymmetry of the frontal and occipital horns of the lateral ventricles. An FDG-PET scan performed at diagnosis showed severe hypometabolism in the left frontal, temporal, and parietal cortices, as well as in the left caudate, putamen, and thalamus nuclei and the right cerebellar cortex (Fig. 2). Plasmatic progranulin levels were normal (30.2 ng/mL). Her 43-year-old daughter is asymptomatic. She decided to undergo genetic testing after genetic counseling, and was found to be a carrier of the variant c.1414-1G>T in the GRN gene.

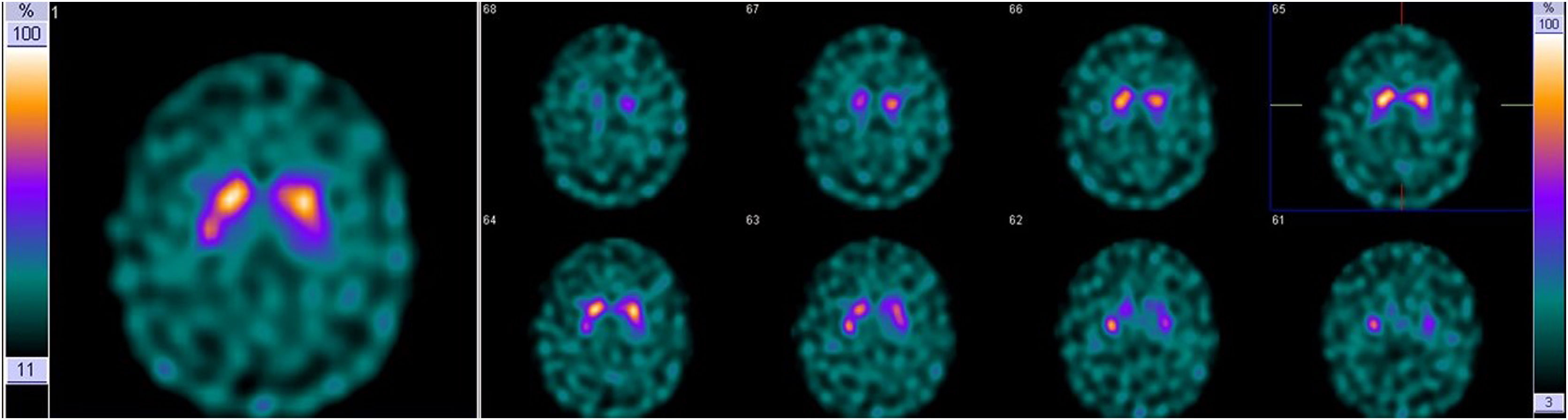
Family #3: The index case is a 62-year-old man with a family history of dementia: his mother and a maternal uncle had been diagnosed with Alzheimer’s disease. His father presented head tremor. The patient started with a resting tremor of the right upper limb. Specifically asked, he admitted presenting hyposmia for at least 3 years. The brain MRI and FDG-PET were both normal, while the DaTSPECT scan revealed a mild decrease, not significant, in the density of presynaptic transporters of dopamine in neurons of the left putamen (Fig. 3). This clinical picture remained stable for 3 years without the use of any dopaminergic medication.

Family #4: The index case is a 57-year-old woman with no relevant medical history. Her family started noticing subtle changes in behavior, including emotional lability. Months later she started with language difficulties and soon later dysphagia and some dyspnea when talking. Then she became clumsy with her right hand. Examination revealed severe dysarthria with no tongue atrophy or fasciculations. Segmentary muscular balance in the limbs was normal. The sensory exam showed no abnormalities. Reflexes were brisk at all levels including bilateral clonus Achilles reflexes. Abdominal reflex and plantar reflexes were negative. Gait was normal. The score in the ALSFRS-R was 41.
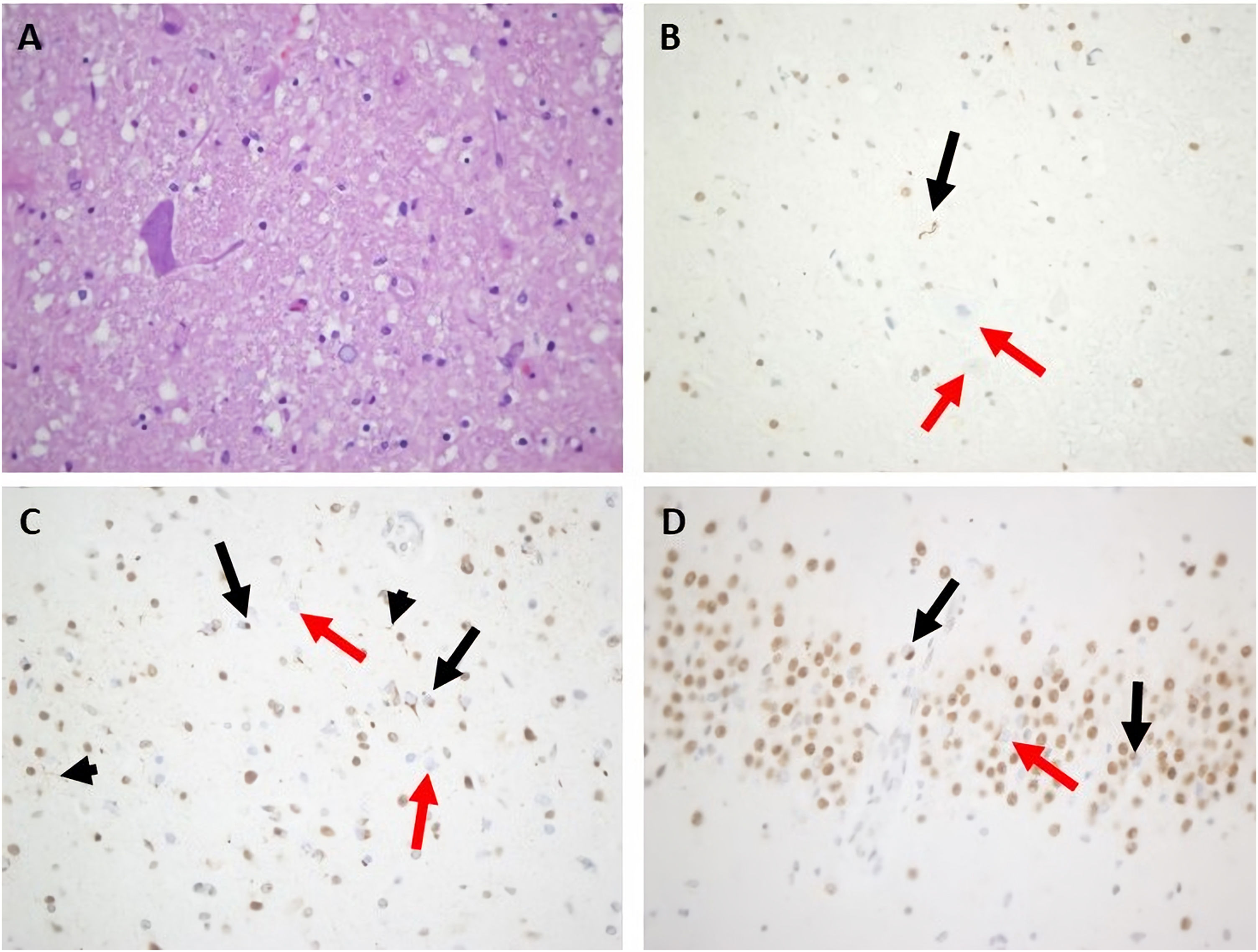
Routine laboratory blood tests were normal. Brain and cervical spine MRI only showed a pineal cyst of 15 mm and a C5-C5 discal hernia without myelopathy. The first EMG was normal but showed mild alterations in the median nerve suggestive of entrapment in the carpal tunnel. The second EMG, 6 months later, showed active denervation signs including spontaneous fasciculations in the bulbar, cervical, thoracic, and lumbar segments, more prominent on the right side, without involvement of the sensory nerves but with bilateral focal neuropathy of the median nerve at the carpal tunnel. Motor-evoked potentials showed altered efferent pathway conduction in the upper limbs only. An FDG-PET scan showed hypometabolism in the frontotemporal lobes, more intense on the left side. Around 12 months after onset, the patient started with painful spasms that were treated with oral levetiracetam, clonazepam, and baclofen, with no improvement. Her mental and motor status deteriorated rapidly, and she died within a few weeks. Autopsy (Fig. 4) revealed findings compatible with FTD-ALS with inclusions of TDP-43 type A. There was an intense loss of motor neurons in the anterior horns in all 3 spinal cord segments, particularly in the cervical and thoracic segments. The remaining neurons showed degenerative signs such as central chromatolysis and were surrounded by areas of gliosis and sclerosis. Immunohistochemistry showed a variable number of TDP-43positive cytoplasmatic inclusions, mostly filiform types. Other motor neurons showed a lack of expression of TDP-43 in the nucleus. Remarkably, there was a moderate-intense mobilization of CD68 positive cells from the microglial-macrophagic system in the white matter of the spinal cord, corresponding to the brainstem corticospinal tracts, displaying an intense loss of the motor neurons in the hyoglossus nuclei, with some neuronophagia. At the encephalic level, conventional staining showed signs of degeneration with neuronal loss in the frontotemporal lobes and micro-spongiosis in the most superficial layers. Immunohistochemistry showed TDP-43–positive intracytoplasmic inclusions, mostly granular and rounded, in the most superficial layers of the frontal cortex (layer 2), with isolated positive short dystrophic neurites. No intranuclear TDP-43–positive inclusions were observed. The hippocampus was scarcely affected, with some intracytoplasmic TDP-43–positive inclusions in the gyrus dentatus. There was no immunoreactivity for AT8 nor beta-amyloid in the cortex or the limbic system. The anti-ubiquitin antibody correlated with the TDP-43–positive inclusions.
 Marked depletion of motor neurons from the anterior horn of the spinal cord (hematoxylin and eosin stain). B) Motor neuron filiform cytoplasmic inclusion was revealed by anti–TDP-43 in the spinal cord (black arrow). Other remaining motor neurons showed loss of normal nuclear staining (red arrows). C) Many small cortical neuronal paranuclear inclusions were detected with antibodies against TDP-43 (black arrows). Short cortical dystrophic neurites were observed with anti–TDP-43 (black arrowheads) Loss of normal nuclear staining of TDP-43 was noted (red arrows). D. Some hippocampal neuronal cytoplasmic inclusions in dentate granule cells were displayed (black arrows). Loss of normal nuclear staining of TDP-43 was noted (red arrow). Magnification ×400.")
Main neuropathological features of index case from family #4. A) Marked depletion of motor neurons from the anterior horn of the spinal cord (hematoxylin and eosin stain). B) Motor neuron filiform cytoplasmic inclusion was revealed by anti–TDP-43 in the spinal cord (black arrow). Other remaining motor neurons showed loss of normal nuclear staining (red arrows). C) Many small cortical neuronal paranuclear inclusions were detected with antibodies against TDP-43 (black arrows). Short cortical dystrophic neurites were observed with anti–TDP-43 (black arrowheads) Loss of normal nuclear staining of TDP-43 was noted (red arrows). D. Some hippocampal neuronal cytoplasmic inclusions in dentate granule cells were displayed (black arrows). Loss of normal nuclear staining of TDP-43 was noted (red arrow). Magnification ×400.
Segregation studies on living family members found that her mother (86 years old, diagnosed with unspecified dementia), a brother (54 years old, asymptomatic), and a sister (66 years old, asymptomatic) were all heterozygous carriers of the c.1414-1G>T variant (Supplementaryfile 2,Fig. 1).
Family #5. Index case (individual II.4 in supplemental file 2, figure 2) is a woman who started with cognitive impairment at 76 years old, mainly memory complaints and language difficulties. The neuropsychological assessment confirmed low scores in naming tasks, comprehension of sentences, and verbal memory. CT was normal while the brain SPECT showed mild-moderate hypoactivity in the frontotemporal regions, more marked in the right hemisphere, and irregularities in the perfusion of parietal lobes (Fig. 5). She progressed slowly and died 8 years later. No specific diagnosis was made, other than “cognitive impairment of probable neurodegenerative origin”. Another member of this family (the younger sister, II.5, Supplementary file 2, Fig. 2) had been assessed independently at another Spanish hospital and diagnosed with Usher-like phenotype, as she presented with late-onset retinal degeneration with very rapid progression and hearing loss, as well as uncharacterized cognitive impairment. She was found to be a homozygous carrier of this variant. We were unable to test all members of this family; but in some cases, the genetic status could be inferred from descendants or known from a previous report.19 The parents of this sibling were a man who had died of lung cancer in his fifties and a woman with visual impairment of no specific diagnosis (not clearly attributable to retinal degeneration). The parents were not tested, but as there were homozygosity among the sibling, both progenitors must have been obligate carriers of the mutation. Two of the mother’s brothers (not shown in the pedigree) had manifested behaviour changes compatible with FTD, although they were not formally diagnosed. The sibling of 6 includes the eldest sister, a heterozygous carrier who died after a stroke and had visual impairment of no specific diagnosis; and a brother (second sibling) who died with unspecified cognitive and visual impairment, whose genetic status is unknown. The younger and elder sisters of the index case (the third and fifth siblings) shared the phenotype of cognitive impairment, hearing loss, and retinal degeneration; and both were found to be homozygous carriers of the variant. Retinal degeneration was compatible with retinitis pigmentosa: age at onset 40-45 years old, with nyctalopia and photophobia; constriction of visual fields evolving to tunnel vision with central vision only distinguishing shapes; fundoscopy revealed thinning of central retinal vessels, a scarce number of bone spicule pigments, absence of foveal reflex, and pale optic disc. Finally, the youngest sister in the sibling (the only one who is still alive) has no symptoms to date but was found to be an asymptomatic carrier. Among the descendants of the sibling, who are now in their fifties and sixties, there are no reported cases of visual or cognitive impairment to date (there is one man with hearing loss, III.3 in Supplementary file 2, Fig. 2). Some of the descendants decided after genetic counseling to get genetic testing done, and some of them resulted to be asymptomatic carriers of the variant 1414-1G > T in GRN gene history (Supplementary file 2, Fig. 2). We measured the levels of plasmatic progranulin in one of the asymptomatic carriers (the son of the index case: III.6 in supplementary file 2, Fig. 2) and resulted within the normal range (37.5 ng/mL).
Discussion
Here we report the main clinical, laboratory, and neuroimaging features of 5 Spanish families with the variant c.1414-1G>T in the GRN gene. This variant was previously described in 2 Hispanic families in the American continent, one of which had 20 affected individuals, but it did not show perfect segregation.20 At that time, the variant c.1414-1G>T in GRN had not been described in other patients or healthy subjects. Later, it was described as part of the spectrum of Usher syndrome in homozygous subjects19 who are also members of family #5 reported here). Given it has only been reported in families of Hispanic origin, and our families are all from the same region, there might be a Spanish founder effect, even though other genetic studies such as haplotype studies would be needed to confirm this point.
Characteristic phenotypic differences corresponding with specific gene variants increase our understanding of the genotype-phenotype relationship in the complex spectrum of neurodegenerative disorders, particularly in familial FTLD, where 3 genes are responsible for most cases.1,2 In our series, information on some families is limited since this is a retrospective study, and information was gathered from medical records and indirectly from other family members. Even so, our series further expands the spectrum of clinical symptoms in GRN mutations. There were homozygous carriers clinically diagnosed with BvFTD, with svPPA, with rapidly progressive motor neuron disease ( pathologically documented), and with tremor dominant parkinsonism; and homozygous carriers diagnosed with retinal degeneration (compatible with retinitis pigmentosa). The latter phenotype was previously reported to be associated with hearing loss in some cases.19 Motor neuron disease is uncommon among carriers of GRN mutations,2 although this association has already been described in a few families,5,21
The most common neuroimaging finding in our series was asymmetric atrophy of the frontal cortex with asymmetric enlargement of the ventricular horns, a well-known characteristic of GRN mutations.22 We also found severe cortical asymmetric hypometabolism with contralateral cerebellar diaschisis and normal DaTSPECT in a patient with tremor parkinsonism, as previously reported in FTD with parkinsonism.23
Clinical heterogeneity is well known in GRN mutations2,5,16,24,25 and there might be different underlying pathological processes explaining this fact. First, GRN expression mediates neuroinflammation function related to general neurodegeneration, which may be a shared mechanism for PD, AD, and ALS.12 In addition, the GRN gene has recently been identified as a susceptibility gene for AD in a large GWAS study,26 which may expand the phenotypic diversity. Interestingly, one of our families is associated with retinitis pigmentosa and it is known that homozygous mutations in GRN are associated with neuronal ceroid lipofuscinosis 11 (CLN11), a rare lysosomal-storage disorder characterized by cerebellar ataxia, seizures, retinitis pigmentosa, and cognitive disorders, usually beginning between 13 and 25 years of age. Other homozygous patients present distinct delayed phenotypes of FTD and parkinsonism after 50 years.27 In the CLN11 gene, hallmarks of neuronal ceroid lipofuscinosis were present, but TDP-43 cytoplasmic inclusions were absent, which markedly differs from heterozygous mutations, suggesting a pathological shift between lysosomal and TDP-43 pathologies depending on the mono- or bi-allelic status. A dosage effect of GRN mutation is supported by the presence of residual levels of plasma PGRN and low levels of normal transcript detected in cases with a homozygous splice-site mutation and late-onset FTD27 and from reports such as the present series, where homozygous carriers developed retinal and brain degeneration while heterozygous carriers developed brain degeneration only or remained asymptomatic.
The neuropathological study of the index case of family 4 shows typical findings of FTD-ALS TDP-43 subtype A,28 which has been reported to be familial in up to 50% of cases. In our series, family history was not particularly informative in some families, with just one or a few members presenting neurodegenerative diseases. Although this suggests incomplete penetrance of this variant, as previously reported,12 the phenotypic heterogeneity of GRN mutations may hinder the identification of family members with related disorders in some cases. Moreover, the age of onset is also variable, with carriers developing severe symptoms in their fifties while others remain asymptomatic in their sixties. It has been shown that both modifier genes29 and epigenetic marks30 contribute to the increased progranulin expression in cases of reduced penetrance.
The variant c.1414-1G>T affects the last nucleotide of intron 11 within a core splice site and is predicted to alter an acceptor splice-site causing exon skipping. Most GRN pathogenic variants are predicted to cause the pathology via a haploinsufficiency mechanism by creating premature stop codons, which in turn result in NMD. We showed that the heterozygous carriers of the variant c.1414-1G>T have a loss of 20 amino acids (468 to 492) in the exon 12 affecting PGRN domain D (granulin D), but no NMD was detected, and the plasmatic levels of the protein were in the normal range according to the laboratory reference.31,32 However, the fact that it is translated into protein does not mean the protein is completely functional. Therefore, functional studies should be performed to assess the functionality of the mutant protein.
Previous works demonstrated that cerebrospinal fluid progranulin levels do not correlate with its serum levels, indicating differential regulation of its central and peripheral levels in neurodegeneration.32 It might be possible that the c.1414-1G>T variant causes a central nervous PGRN reduction or has a pathological mechanism similar to other pathogenic variants associated with normal progranulin levels.33 Future studies are needed to determine these possibilities.
A recent study34 showed that affected pathways in GRN mutation carriers extend beyond GRN and contribute to genetically unexplained forms of FTLD-TDP type A, including dysregulation of the GABAergic signaling pathway and the potential involvement of astrocytic inflammation. This study also provides new avenues for research into FTLD-TDP type A and GRN and potentially new therapeutic targets such as the GABAergic, GDNF, and sphingolipid pathways. Other therapeutic strategies such as gene therapy targeting splicing sites may also be suitable.
In conclusion, the clinical pictures, segregation data, the absence of variant carriers among non-demented controls, and splicing analyses strongly suggest that GRN c.1414-1G>T is a pathogenic variant. Given it has only been reported in families of Hispanic origin, and our families are all from the same region, there might be a Hispanic founder effect. As this variant seems to affect GRN gene splicing but not plasmatic levels of the progranulin protein, there might be other pathogenic mechanisms apart from haploinsufficiency; functional studies are needed to elucidate this question. Phenotypes and disease progression can be diverse as in other GRN mutations, while retinitis pigmentosa may be added to the clinical spectrum. This variant should be considered for therapeutic strategies based on the GABAergic, GDNF, lysosomal function, and sphingolipid pathways or for those based on restoring GRN functional levels, either using enzymatic inhibition or using gene editing targeting splicing sites.35–37
Competing interestsThe authors report no competing interests.
FundingThis research was funded by a grant from Instituto de Salud Carlos III (PI21/00467, co-funded by European Regional Development Fund/European Social Fund) to MMG and VA.
We would like to thank patients and families for collaborating in this study and giving permission to publish the series of cases.
The following are Supplementary data to this article:
