




Charcot-Marie-Tooth disease (CMT) is classified according to neurophysiological and histological findings, the inheritance pattern, and the underlying genetic defect. The objective of these guidelines is to offer recommendations for the diagnosis, prognosis, follow-up, and treatment of this disease in Spain.
Material and methodsThese consensus guidelines were developed through collaboration by a multidisciplinary panel encompassing a broad group of experts on the subject, including neurologists, paediatric neurologists, geneticists, physiatrists, and orthopaedic surgeons.
RecommendationsThe diagnosis of CMT is clinical, with patients usually presenting a common or classical phenotype. Clinical assessment should be followed by an appropriate neurophysiological study; specific recommendations are established for the parameters that should be included. Genetic diagnosis should be approached sequentially; once PMP22 duplication has been ruled out, if appropriate, a next-generation sequencing study should be considered, taking into account the limitations of the available techniques. To date, no pharmacological disease-modifying treatment is available, but symptomatic management, guided by a multidiciplinary team, is important, as is proper rehabilitation and orthopaedic management. The latter should be initiated early to identify and improve the patient’s functional deficits, and should include individualised exercise guidelines, orthotic adaptation, and assessment of conservative surgeries such as tendon transfer. The follow-up of patients with CMT is exclusively clinical, and ancillary testing is not necessary in routine clinical practice.
La enfermedad de Charcot-Marie-Tooth (CMT) se clasifica según las características neurofisiológicas e histológicas, el patrón de herencia y el defecto genético subyacente. El objetivo de esta guía es establecer recomendaciones prácticas para el diagnóstico, pronóstico, seguimiento y tratamiento de esta enfermedad en España.
Material y métodosSe trata de un proyecto colaborativo y multidisciplinar contando con un grupo amplio de profesionales expertos en la materia e incluyendo neurólogos, neuropediatras, neurofisiólogos, genetistas, rehabilitadores y cirujanos ortopédicos.
RecomendacionesEl diagnóstico de sospecha en CMT es clínico, habitualmente detectando un fenotipo común o clásico. La evaluación clínica se debe seguir de un estudio neurofisiológico adecuado y se establecen recomendaciones concretas sobre los parámetros que deben ser recogidos. El diagnóstico genético debe abordarse secuencialmente; una vez descartada la duplicación del gen PMP22 si corresponde, se recomienda realizar un estudio de secuenciación masiva teniendo en cuenta las limitaciones de las técnicas. No existe tratamiento farmacológico modificador del curso de la enfermedad, si bien es importante el manejo sintomático guiado por un equipo multidisciplinar, así como el adecuado abordaje rehabilitador y ortopédico. Éste debe iniciarse precozmente para identificar y mejorar los déficits funcionales del paciente e incluye pautas individualizadas de ejercicio, adaptación ortésica y valoración de cirugías conservadoras como la transposición de tendones. El seguimiento de los pacientes es clínico, no siendo necesario la realización de pruebas complementarias en la práctica clínica habitual.
Hereditary motor and sensory neuropathy, also known as Charcot-Marie-Tooth disease (CMT), is the most frequent form of hereditary neuropathy.1 The classical phenotype is a length-dependent motor and sensory neuropathy characterised by distal limb weakness, sensory alterations, loss of reflexes, and pes cavus. Our knowledge of this genetically heterogeneous entity has increased with the development of next-generation genetic sequencing techniques; at present, over 100 genes are known to be associated with the disease.2 Several pathogenic variants have been described in genes encoding proteins with highly variable function and localisation (myelin, Schwann cells, axons, etc), although all of them ultimately result in axonal degeneration.
CMT is classified according to neurophysiological and/or histological findings, inheritance pattern, and the underlying genetic defect. Thus, we may differentiate between demyelinating forms, in which motor nerve conduction velocity (NCV) in the upper limbs is < 38 m/s (known as CMT1 if an autosomal dominant pattern is observed and CMT4 if the pattern of inheritance is autosomal recessive), and axonal forms, in which motor NCV in the upper limbs is > 38 m/s (known as CMT2, regardless of the inheritance pattern).3 An intermediate subtype is also recognised, with somewhat less precise motor NCV values (initially established at 25-45 m/s, although subsequent studies have established cutoff values between 30-40 m/s); this subtype is frequently associated with an X-linked inheritance pattern (CMTX), although cases of autosomal dominant or autosomal recessive transmission have also been identified.4 Demyelinating, axonal, and intermediate forms may also be classified according to the causal genetic defect, with the most frequent subtype being CMT1A, caused by a duplication affecting the PMP22 gene.2
The Spanish Society of Neurology’s Neuromuscular Diseases Study Group created this consensus document on the diagnosis and management of patients with CMT.
MethodsThis collaborative, multidisciplinary project involved a large group of professionals representing all specialties participating in the management of CMT (neurology, neuropaediatrics, neurophysiology, clinical genetics, rehabilitation medicine, orthopaedic surgery, etc). The project also sought to take into account patients’ perspectives and preferences.
We conducted a literature search of the PubMed-MEDLINE, EMBASE, ECA LOST, Cochrane Library, and Cochrane Plus databases, following the PICO (patient, intervention, comparison, outcome) method whenever possible, as recommended by the working group for clinical practice guidelines of the Spanish National Healthcare System.5 We used the search terms “Charcot Marie Tooth disease,” “CMT,” and “hereditary neuropathy,” specifying no date range. We selected the most recent and highest-quality references found in the literature.
Based on the reviewed articles, this document was developed by different working groups, each focusing on one of the following topics: introduction and methods, diagnosis (clinical, neurophysiological, genetic, and other ancillary tests), genetic counselling, treatment (pharmacological, rehabilitation, orthopaedic, and disease-modifying), and follow-up. Each group presented a document, which was discussed and agreed upon with the remaining participants over the course of several meetings, to establish consensus on the recommendations expressed in the present guidelines. Subsequently, the initial manuscript was drafted and was reviewed on multiple occasions by all authors, with the conclusions being approved once more in another meeting. Finally, the AGREE II tool was used to evaluate and guarantee the quality, clarity, rigour, applicability, and editorial independence of these guidelines.6
During the drafting of this document, we discussed the challenges and potential costs associated with the application of these recommendations. All authors approved the final version of this manuscript and take full responsibility for its content.
DiagnosisClinical diagnosis — classical phenotypeThe first step in the diagnosis of CMT is to determine whether the patient presents a hereditary neuropathy. Several non-specific findings may suggest CMT: 1) family history of the disease; 2) childhood onset; 3) slowly progressive course; 4) presence of skeletal deformities, such as pes cavus; and 5) scarcity of positive sensory symptoms (paraesthesia or dysaesthesia), despite clear sensory deficits.7
Despite the great genetic heterogeneity of the disease, patients with CMT may present a common or classical phenotype. Symptoms typically appear in the first or second decade of life, with CMT1 frequently presenting at younger ages than CMT2.8 The initial manifestations frequently involve the lower limbs, with weakness and progressive muscle atrophy of the distal muscles. This results in difficulty running and toe and heel walking, as well as frequent falls. Over the years, the disease typically progresses to affect the upper limbs, leading to problems with fine motor skills (eg, writing, buttoning a shirt). Weakness and atrophy of the intrinsic muscles of the foot cause deformities such as pes cavus, claw toes, and Achilles tendon retraction. Pes cavus is a key manifestation of the disease that indicates that the process of denervation started in childhood, although during the first 2 years the predominant sign may be hypotonic, flat valgus foot.9,10
As the disease progresses, the distal muscles of the legs and those in the lower third of the thigh may present atrophy, giving the appearance of a stork’s leg or a reversed champagne bottle. These patients may also display sensory symptoms, mainly alterations in tactile and vibration sensitivity. Positive sensory symptoms are rarely reported. Muscle stretch reflexes may be reduced or absent, particularly in the lower limbs. A rather specific sign of such demyelinating forms as CMT1A is the thickening of the peripheral nerves, which can be felt or even seen through the skin.7 Less frequently, patients may present cranial neuropathies, tremor, scoliosis, muscle cramps, and contractures.
The disease frequently presents a slowly progressive course. The vast majority of patients are independent and maintain the ability to walk autonomously for much of their lives, although some present severe subtypes associated with significant functional impairment. On the other hand, clinical variability may be observed both between and within families, even within the same genotype.
Clinical diagnosis — CMT subtypesThe clinical characteristics of the most frequent subtypes are presented below; Supplementary material 1 summarises the most relevant clinical characteristics of other subtypes. Overall, the most frequent genetic subtypes are those caused by a duplication that includes the PMP22 gene (CMT1A), followed by pathogenic point mutations in the genes GJB1 (CMTX1), MPZ (CMT1B), MFN2 (CMT2A), and GDAP1 (CMT4A, CMT2K).11,12
CMT1A: This is the most common subtype of CMT, representing 40%-50% of all cases and 60%-70% of CMT1.11 It is caused by a 1.5-Mb duplication in the chromosome region 17p11.2-p12, where the PMP22 gene is located. This results in overexpression of PMP22 and accumulation of the protein in Schwann cells, causing cell damage. Most patients with CMT1A present the classic phenotype.13 Forms caused by pathogenic variants of the same gene (CMT1E) are much less frequent, and their phenotype is variable, ranging from severe early-onset forms to milder late-onset forms.14
CMT1B: This subtype is caused by variants in the MPZ/P0 gene, representing approximately 8% of all cases of CMT. Most changes in this gene cause a demyelinating phenotype (CMT1B) similar to CMT1A or severe childhood forms manifesting with motor developmental delay and a congenital hypomyelinating neuropathy phenotype.15,16 These variants may also cause an axonal phenotype (CMT2I/J), with later onset.15,17
CMT2: This group presents great genetic heterogeneity, resulting in significant clinical variability (Supplementary material 1), although many subtypes share a classical phenotype. The most common subtype globally is CMT2A, caused by changes in MFN2, although in Spain there is a high prevalence of patients with dominant pathogenic variants in the GDAP1 gene (CMT2K).18 As a whole, asymmetry is more common than in patients with demyelinating forms (20%).19,20
CMTX: This subtype includes X-linked forms of CMT, which may be dominant (more frequently) or recessive. CMTX1 is the most common subtype, caused by pathogenic variants in the GJB1 gene, and represents the second most frequent form of CMT (7%–12%). Given its X-linked inheritance pattern, men usually present more severe forms and earlier onset than women, who may be asymptomatic or nearly asymptomatic. NCV are usually in the intermediate range, and patients may present clinical and neurophysiological asymmetries.21 CNS involvement is rarely described, and is characterised by transient episodes of focal neurological signs, “stroke-like” symptoms, or encephalopathy, which may precede the diagnosis of peripheral neuropathy.22
Key concepts:
- •
The initial diagnosis of CMT is primarily clinical and should involve the exclusion of other neuropathies or clinically similar neuromuscular diseases.
- •
Despite the great genetic heterogeneity of CMT, patients often display a common or classic phenotype, although some genetic subtypes present distinguishing features.
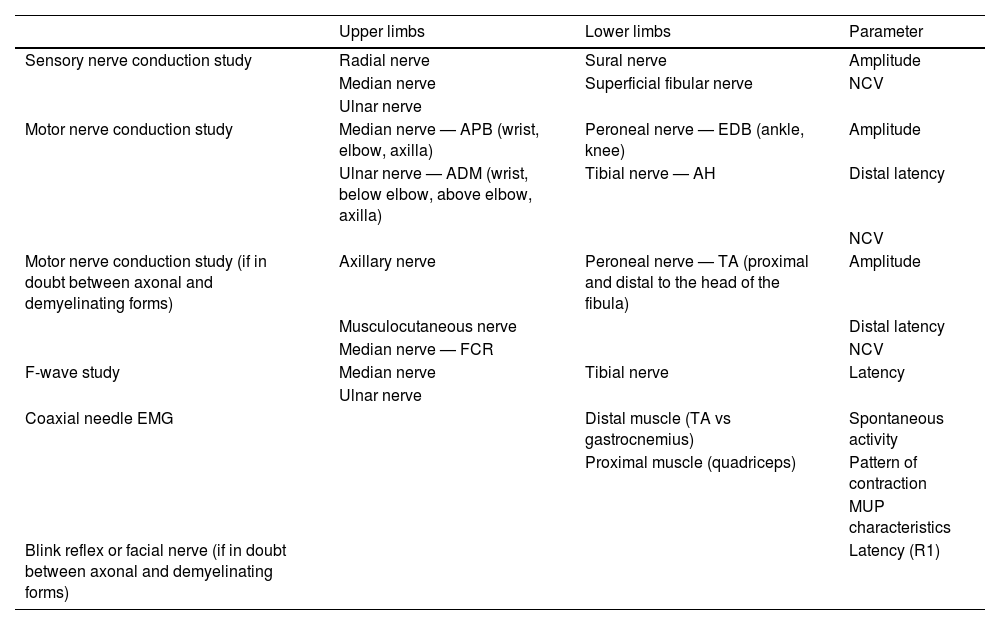
Nerve conduction studies provide relevant data for the diagnosis, classification, and assessment of the pathophysiological mechanisms underlying CMT. It is important to conduct a detailed neurophysiological study that includes both sensory and motor nerves in the upper and lower limbs, examining both proximal and distal nerve segments. Furthermore, taking into account the amplitude of motor responses is crucial for the correct interpretation of NCV. The parameters recommended for the neurophysiological study of these patients are listed in Table 1.4,7
Recommended protocol for the neurophysiological study of Charcot-Marie-Tooth disease.
| Upper limbs | Lower limbs | Parameter | |
|---|---|---|---|
| Sensory nerve conduction study | Radial nerve | Sural nerve | Amplitude |
| Median nerve | Superficial fibular nerve | NCV | |
| Ulnar nerve | |||
| Motor nerve conduction study | Median nerve — APB (wrist, elbow, axilla) | Peroneal nerve — EDB (ankle, knee) | Amplitude |
| Ulnar nerve — ADM (wrist, below elbow, above elbow, axilla) | Tibial nerve — AH | Distal latency | |
| NCV | |||
| Motor nerve conduction study (if in doubt between axonal and demyelinating forms) | Axillary nerve | Peroneal nerve — TA (proximal and distal to the head of the fibula) | Amplitude |
| Musculocutaneous nerve | Distal latency | ||
| Median nerve — FCR | NCV | ||
| F-wave study | Median nerve | Tibial nerve | Latency |
| Ulnar nerve | |||
| Coaxial needle EMG | Distal muscle (TA vs gastrocnemius) | Spontaneous activity | |
| Proximal muscle (quadriceps) | Pattern of contraction | ||
| MUP characteristics | |||
| Blink reflex or facial nerve (if in doubt between axonal and demyelinating forms) | Latency (R1) |
ADM: abductor digiti minimi; AH: abductor hallucis; APB: abductor pollicis brevis; EDB: extensor digitorum brevis; EMG: electromyography; FCR: flexor carpi radialis; MUP: motor unit potential; NCV: nerve conduction velocity; TA: tibialis anterior.
In the demyelinating subtype, neurophysiological studies have shown uniform, marked decreases in motor and sensory NCV across the nerves in the upper and lower limbs, with marked increases in distal latencies and prolonged or abolished F-wave latencies. The accepted cut-off point for motor NCV in upper limb nerves is < 38 m/s. The amplitude of motor and sensory nerve action potentials decreases with age and disease progression. In fact, clinical severity largely depends on secondary axonal damage, rather than on the magnitude of the decrease in NCV.7
Hereditary demyelinating neuropathies have traditionally been considered to cause a uniform decrease in NCV along the nerve, across all nerves, whereas acquired demyelinating neuropathies present more irregular involvement, with focal slowing along the trajectory of the nerve, varying across nerves, as well as conduction blocks and/or temporal dispersion. However, there are exceptions to this rule, and increasing numbers of genes responsible for demyelinating CMT have been reported to cause conduction blocks and/or temporal dispersion (GJB1, MPZ, SH3TC2, SPTLC1, FIG4, PMP22).23,24 In CMT1A, mean motor NCV is 21 m/s, whereas in severe or recessive forms of demyelinating CMT it is usually much slower (4-15 m/s).25 Prolonged distal motor latency is frequently the initial manifestation of demyelination and may occur at ages as young as one year, before NCV slowing is observed, usually at 3-5 years of age.26 In cases of severe demyelination and markedly decreased amplitude or absence of motor response, increased latency of the blink reflex (R1 > 13 ms) or of the facial nerve demonstrates a good correlation with hereditary demyelinating neuropathies.23
In the axonal subtype, conduction studies reveal low potential amplitudes and normal conduction velocities, both in sensory and motor nerves (motor NCV in upper limb nerves > 38 m/s).7 In advanced stages of axonal degeneration, distal velocities may be slowed due to loss of large myelinated fibres. In cases in which the distal compound motor action potential presents a significantly decreased amplitude and it is unclear whether involvement is axonal or demyelinating, we recommend studying conduction to more proximal muscles, testing the blink reflex, or evaluating latencies of short nerves such as the axillary or musculocutaneous nerves (which present normal values in the event of axonal involvement).
Nerve conduction studies are useful for distinguishing CMT2 from distal hereditary motor neuropathies (dHMN), as the latter present normal sensory conduction. It is now known that these 2 entities represent a pathological continuum, given that there are certain genes, like HSPB1, which may cause both CMT2 and dHMN phenotypes.25 The same is true for sensory forms, in which motor conduction is initially preserved.27
In both subtypes, needle EMG reveals signs of chronic denervation, with a pattern of reduced recruitment, increased amplitude and prolonged duration of motor unit potentials, and polyphasia, predominantly affecting distal limb muscles. Other findings include signs of active denervation, such as fibrillation potentials and positive waves.
The term intermediate CMT is somewhat controversial, and should only be used to describe a form of CMT, rather than to refer to isolated NCV values in a single nerve.28 To define this pattern, the limits of motor NCV were initially established at 25-45 m/s, although this range was later narrowed to 30-40 m/s.4 The histopathological findings of the first series of intermediate CMT reveal a combination of demyelinating and axonal changes, which would explain this mixed pattern. To accurately classify the condition, motor nerve conduction studies should be performed in proximal nerve segments, including conduction to proximal muscles.4
Key concepts:
- •
Nerve conduction studies provide relevant data for the diagnosis, classification, and prognosis of patients with CMT.
- •
Nerve conduction studies are recommended in all patients with clinical suspicion of CMT, unless there is a contraindication.
- •
Nerve conduction studies should test proximal and distal segments of sensory and motor nerves of the upper and lower limbs, as shown in Table 1.
- •
Correct interpretation of NCV must take into consideration potential amplitudes.
CMT is a genetically heterogeneous disorder. For an accurate genetic diagnosis, it is essential to establish beforehand the clinical phenotype, electrophysiological characteristics, and the possible pattern of inheritance. For this reason, the clinical evaluation of a patient with CMT should include obtaining a detailed family tree of at least 3 generations. It should be noted that the absence of clear family history does not rule out hereditary neuropathy. It is also important to consider the prevalence of different genetic subtypes of CMT in the patient’s population and ethnic group.11,12 For example, in patients of Romani ethnicity with CMT1, priority should be given to searching for founder point variants in the SH3TC2, HK1, and NDRG1 genes. Finally, we should also consider other differential diagnoses or even a clinical overlap with such hereditary neuropathies as hereditary neuropathy with pressure palsies (HNPP); distal hereditary motor neuropathies (dHMN); hereditary sensory neuropathies (HSN), or hereditary sensory and autonomic neuropathies (HSAN); disorders within the spectrum of cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS); systemic hereditary diseases presenting predominantly with peripheral neuropathy (eg, familial amyloid neuropathies or neurometabolic diseases); and distal myopathies.
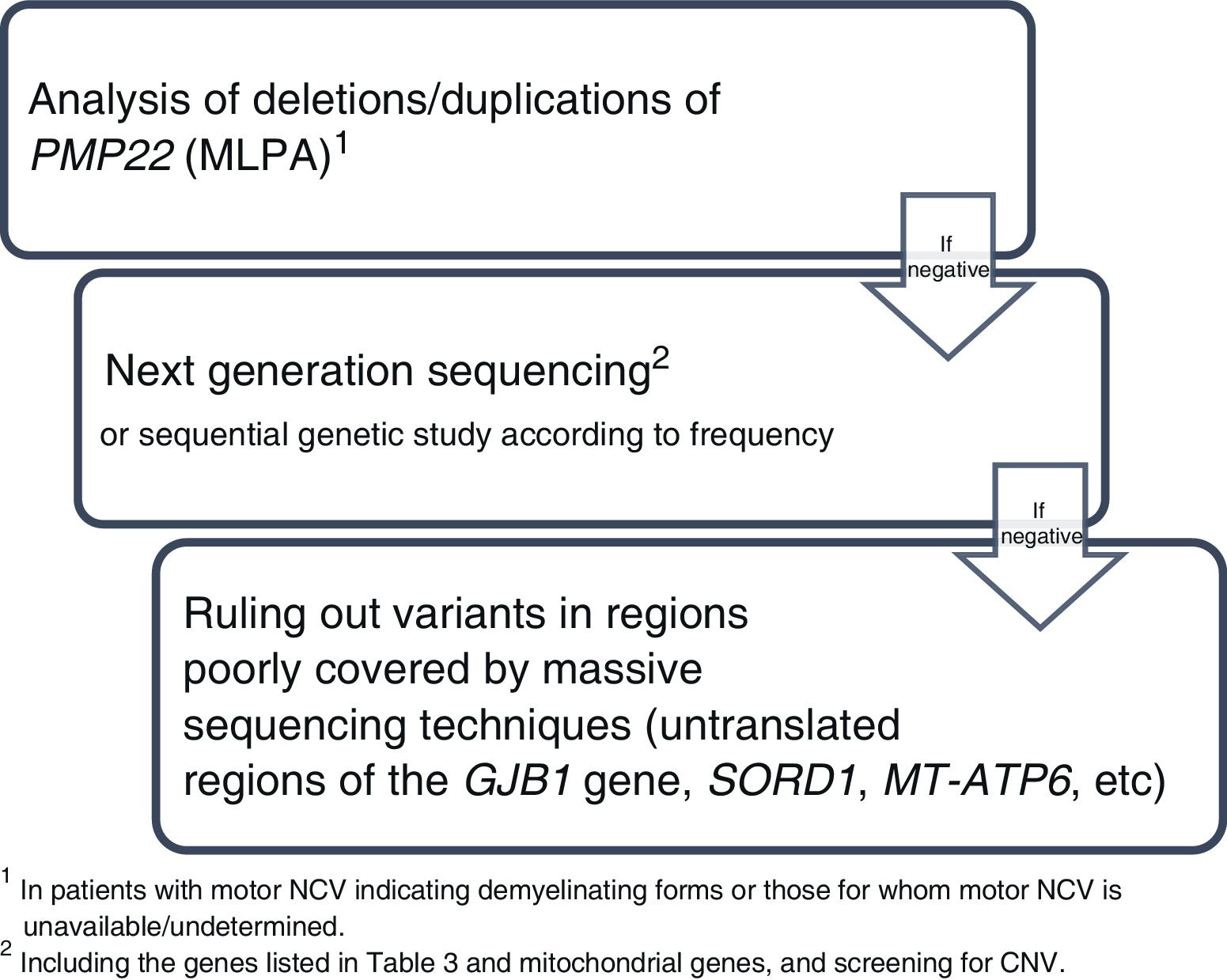
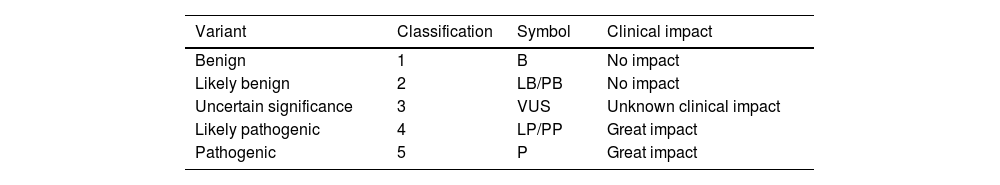
The wide range of diagnostic tests currently available has simplified algorithms for genetic diagnosis and reduced diagnostic delays. Genetic variants are classified according to international consensus standards into 5 classes (Table 2),29 and are described using an international nomenclature (http://varnomen.hgvs.org). Genetic study findings should preferably be interpreted by multidisciplinary committees including neurologists, geneticists, and other specialists; this would enable analysis of the genotype-phenotype correlation of the identified variants and follow-up of the most complex cases. Segregation and/or functional studies may occasionally be necessary to confirm the pathogenicity of a candidate genetic variant. Fig. 1 summarises the recommended sequence for the genetic study of patients with suspected CMT, which is explained in greater detail below.
Classification of variants according to the criteria established by the American College of Medical Genetics and Genomics.
| Variant | Classification | Symbol | Clinical impact |
|---|---|---|---|
| Benign | 1 | B | No impact |
| Likely benign | 2 | LB/PB | No impact |
| Uncertain significance | 3 | VUS | Unknown clinical impact |
| Likely pathogenic | 4 | LP/PP | Great impact |
| Pathogenic | 5 | P | Great impact |
VUS: variant of uncertain significance.

Step 1. Analysis of deletions/duplications of PMP22 (eg, multiplex ligation-dependent probe amplification [MLPA]) in all patients with suspected CMT and at least one of the following characteristics: 1) motor NCV in the median/ulnar nerve of < 38 m/s; 2) unavailable/undetermined motor NCV in the median/ulnar nerve; and 3) patients in whom a neurophysiological study cannot be performed.
Step 2. Once PMP22 duplication is ruled out (step 1), next-generation sequencing techniques should be performed, where available. Gene panel or exome sequencing techniques are typically used, and should include at least the genes most frequently associated with CMT (GJB1, MPZ, PMP22, SH3TC2, MFN2, GDAP1, MME, and HSPB1). Given the frequency of pathogenic point variants in GJB1, direct sequencing of this gene may be performed before massive sequencing in patients with compatible phenotypes (eg, lack of male-to-male transmission, median/ulnar nerve motor NCV in the axonal or intermediate range). Furthermore, given the clinical and genetic overlap between CMT and neuropathies such as dHMN and HSN/HSAN, genetic studies should also include the genes associated with these diseases, as well as genes associated with differential diagnoses (Table 3). Data obtained through next-generation sequencing may be analysed sequentially: the genes associated with CMT should be examined first; if the results are negative, data are analysed from the genes associated with dHMN and/or HSN/HSAN, and finally from genes related to differential diagnoses. This approach minimises incidental findings and variants of uncertain significance, which, due to their unknown clinical impact, tend to have little clinical utility. If these techniques are not available, direct sequencing of individual genes associated with CMT may be performed sequentially.30
Genes that should be analysed through massive sequencing.
| CMT (71 genes) | AARS1/AARS, AIFM1, ARHGEF10, ATP1A1, COX6A1, DCAF8, DHTKD1, DNAJB2, DNM2, DRP2, DYNC1H1, EGR2, FBLN5, FGD4, FIG4, GAN/GAN1, GANP/MCM3AP, GARS1, GBF1, GDAP1, GJB1, GJB3, GNB4, HARS1, HINT1, HK1, HSPB1, HSPB8/HSP22, IGHMBP2, INF2, ITPR3, JPH1, KARS, KIF1B, KIF5A, LITAF, LMNA, LRSAM1, MARS/MARS1, MED25, MFN2, MME, MORC2, MPV17, MPZ, MTMR2, NAGLU, NDRG1, NEFH, NEFL, PDK3, PDXK, PLEKHG5, PMP2, PMP22, PRPS1, PRX, RAB7A, SBF1, SBF2, SH3TC2, SIGMAR1, SORD, SPG11, SURF1, TFG, TRIM2/KIAA0517, TRPV4, TUBB3, VCP, YARS |
| dHMN (20 genes) | BAG3, BSCL2, DCTN1, DNMT1, FBXO38, GARS1, HSPB1, HSPB3, HSPB8/HSP22, IGHMBP2, OPA3, PLEKHG5, REEP1, SETX, SIGMAR1, SLC5A7, TBCK, TRPV4, TTR, WARS1 |
| HSN/HSAN (23 genes) | ATL1, ATL3, DNMT1, DST, ELP1/IKBKAP, HSAN1B (3p24-p22), KIF1A, NGFB/NGF, NTRK1, OPA3, PRDM12, RAB7A, RETREG1/FAM134B, SACS, SAMD9L, SCN10A, SCN11A, SCN9A, SPTLC1, SPTLC2, TECPR2, TRPA1, WNK1 |
| Differential diagnosis (10 genes) | ABCA1, APTX, ELP1/IKBKAP, HMBS, PPOX, RFC1, SETP9, SLC25A46, VRK1, VWA1 |
Step 3: If the results of the previous studies are negative, the following options and considerations will be taken into account:
- •
In the analysis of the GJB1 gene, pathogenic variants have been described in the gene’s regulatory region, in the 5′ and 3′ untranslated regions (5′-UTR and 3′-UTR, respectively), and in the splice site region between exons 1 and 2.31 As these regions are often poorly covered by next-generation sequencing techniques, direct sequencing of the GJB1 gene should be considered.
- •
Pathogenic point variants in the SORD gene, mainly the recurrent variant c.757delG, are reported to be responsible for up to 10% of all cases of autosomal recessive dHMN and CMT2.32SORD is often poorly covered by next-generation sequencing techniques due to the presence of the pseudogene SORD2P. Therefore, other techniques need to be used for its detection.
- •
Pathogenic point variants in the mitochondrial gene MT-ATP6 (eg, m.9185C>T) have been reported to be responsible for at least 1% of all cases of axonal CMT in some series.33 Although the study of mitochondrial DNA is increasingly common in massive sequencing techniques, we must ensure that this possibility is considered. If mitochondrial DNA is not included, direct sequencing techniques may be necessary for detection of these variants.
- •
In addition to the PMP22 gene, copy number variants (CNV) have been detected in other genes, including some of those most frequently associated with CMT, such as GJB1, MPZ, MFN2, and NDRG1.34 Therefore, analysis of this gene should be considered either through direct tests for detecting deletions/duplications (eg, MLPA) or by ensuring that the next-generation sequencing technique used is capable of screening for CNVs in the genes under study.
Key concepts:
- •
All patients with a clinical diagnosis of CMT should be offered a genetic study to establish a molecular diagnosis.
- •
The evaluation of a patient with CMT should include obtaining a detailed family pedigree covering at least 3 generations.
- •
We recommend that genetic testing be conducted sequentially:
- 1
Duplication of the PMP22 gene in patients with demyelinating or undetermined forms.
- 2
Next-generation sequencing study including genes associated with CMT and related diseases.
- 3
Consider complete sequencing of GJB1, SORD, MT-ATP6, etc, as well as CNV analysis in other genes.
- 1
Certain ancillary tests can provide useful information for the diagnosis and/or prognosis of a patient with CMT, although they are not essential in all cases.
Magnetic resonance (MR) neurography provides images of the peripheral nerves and nerve roots, revealing an increase in thickness and intraepineurial fat in patients with CMT, especially those with demyelinating forms.35,36 On the other hand, muscle MRI reveals atrophy and fatty infiltration, mainly affecting the distal muscles of the limbs and correlating with weakness. Muscle MRI protocols should include images of the foot muscles. Studies published in recent years have described patterns of fatty infiltration that could be suggestive of certain subtypes of CMT, although they need to be better defined.37 Furthermore, fatty infiltration of muscles, calculated using quantitative techniques (such as the Dixon method) or semiquantitative methods (such as visual scales), can be useful as a marker of progression.38,39 Likewise, brain MRI can be useful in some subtypes of CMT (MFN2, GJB1, NEFL, etc), where CNS abnormalities can occasionally be observed.22,40
High-resolution ultrasound enables non-invasive morphological assessment of peripheral nerves and cervical nerve roots, and proves useful in distinguishing demyelinating forms, where an increase in the cross-sectional area of all nerves is observed, from axonal forms, in which nerves may present alterations in echotexture without evidence of thickening.41,42 This painless test can also be used in children, for screening relatives of affected individuals, in the diagnosis of overlapping compressive neuropathies, and in differential diagnosis to rule out acquired neuropathies such as chronic inflammatory demyelinating polyneuropathy. Patients with the latter condition display a somewhat less marked nerve thickening, which is most notably asymmetric, focal, and predominantly proximal.43,44
Serum creatine kinase (CK) is a nonspecific marker; levels may be moderately elevated in some patients with CMT, especially in certain predominantly motor forms.45 In any case, routine CK determination is not recommended in patients with suspected CMT. Lumbar puncture is an invasive procedure of limited value in CMT; its use is discouraged except when necessary for differential diagnosis with other conditions such as CIDP. Elevated protein levels (up to 100 mg/dL) have been reported in some patients with CMT. Finally, histological studies now play a secondary role due to the development of molecular diagnostic techniques. Peripheral nerve biopsy is recommended only when a treatable neuropathy (amyloidosis, vasculitis, etc) is suspected and, occasionally, in the event of diagnostic uncertainty or for research purposes.46 Muscle biopsy may be useful for differential diagnosis with a distal myopathy or if copresence of a myopathic and neuropathic disorder is suspected.
Key concepts:
- •
Ancillary tests (muscle MRI, MR neurography, nerve ultrasound, CK determination, histological study, etc) may provide useful information for the diagnosis and/or prognosis of CMT.
- •
These techniques should be indicated at specialised centres, when diagnosis is uncertain, to better profile the patient’s phenotype or for research purposes.
Diagnosis should be disclosed in simple, clear terms within the privacy of the consultation. Sufficient time should be allocated for the patient to understand the nature of their condition and to answer any questions that may arise (see example in Supplementary material 2). The Spanish Royal Decree 1030/2006 establishes that genetic counselling is a fundamental part of the care provided to any patient with a genetic disease, including CMT, and should therefore be available to all patients and families with suspected CMT. It should be provided by qualified personnel, either a geneticist with expertise in the disease or the neurologist themselves. Pre-test genetic counselling should address matters related to the risks and benefits of undergoing genetic testing and the significance of the possible test results.
Post-test genetic counselling involves explaining the hereditary nature of the disease, the possibility that other family members may be affected/carriers, and the risk of transmitting the disease to offspring. A confirmatory genetic study allows for predictive genetic testing (of adult direct relatives who are asymptomatic but are at risk of developing the disease) and for genetic studies of offspring to be performed. The reproductive options covered by the public services portfolio, as established in the Royal Decree, should be discussed with each patient. Reproductive options include both assisted reproduction techniques (e.g., preimplantation diagnosis, gamete or embryo donation, etc) and prenatal diagnosis during pregnancy.47 Predictive studies of adult-onset diseases in minors should be delayed until the individual is sufficiently mature and competent to understand the implications of genetic testing, unless effective preventive measures exist that may be applied in childhood.
Post-test genetic counselling should also be provided even when molecular study results are negative, to explain the possible inheritance pattern and the family members potentially at risk. In these cases, we recommend advising a clinical evaluation of other family members and inform patients that having disease-free offspring is only possible through gamete or embryo donation.
Lastly, genetic counselling should also take into consideration psychosocial issues arising after the diagnosis of a chronic and progressive hereditary disease.48 Therefore, the multidisciplinary team should include a psychologist, and patients and their families should be informed about patients’ associations (eg, ASEM Federation [https://www.asem-esp.org] or FEDER [https://www.enfermedades-raras.org]).
Key concepts:
- •
Diagnosis should be disclosed in clear, simple terms, dedicating sufficient time. It will include appropriate individual, family, and reproductive genetic counselling by qualified personnel.
- •
Genetic counselling should be provided both before and after the genetic study, even if the results are negative.
- •
A confirmatory genetic study enables the performance of predictive genetic testing in adult direct relatives at risk of developing the disease and the planning of reproductive options to avoid transmitting the disease to offspring.
- •
As established in the Spanish Royal Decree 1030/2006, predictive genetic testing is not recommended in children and adolescents.
No disease-modifying treatment is currently available for CMT, although proper management of symptoms is essential. This may include pharmacological treatments, rehabilitation, and orthopaedic strategies. A multidisciplinary approach, adapted to each patient’s characteristics, is essential.
Symptomatic treatmentPatients with CMT frequently report pain, fatigue, and cramps; evaluation and treatment of these symptoms is essential to improve patients’ quality of life. Pain is reported by 23%-85% of patients, and may be of biomechanical or neuropathic origin.49 Neuropathic pain should be managed according to the available evidence, mainly using antidepressants (amitriptyline, duloxetine, etc), antiepileptic drugs (gabapentin, pregabalin, carbamazepine, etc), and local anaesthetics (capsaicin, etc), and avoiding opioids.50 Fatigue is also a frequent symptom in patients with CMT. No specific pharmacological treatment for fatigue is currently available; management strategies focus on related aspects, including somnolence, obesity, and anxiety.51 Finally, cramps may be present in up to 85% of patients and should be managed similarly to those of any other aetiology, as there are no specific recommendations.52
Routine administration of vitamin complexes is not recommended unless there is evidence of a nutritional deficiency, as the available evidence suggests that vitamin C supplementation does not improve the course of CMT1A.53,54 No conclusive data are available for recommending specific diets or supplements, although it is good practice to promote global health measures including primary prevention of obesity and diabetes.
Depending on the genetic subtype and clinical severity of CMT, it is advisable to evaluate the presence of other associated signs and symptoms. An assessment should be conducted to detect respiratory failure in severe forms of the disease, associated with scoliosis, or to identify suggestive symptoms.55 Furthermore, sleep disorders such as sleep apnoea/hypopnoea syndrome (SAHS) and restless legs syndrome are highly prevalent among these patients; targeted history-taking is recommended, and a polysomnography study may be performed when necessary.56 No specific recommendations are currently available for the management of either condition. Cranial nerve involvement is common in some subtypes of CMT and may cause disorders such as sensorineural hearing loss, vocal cord paralysis, and optic neuropathy, which should be assessed and treated by appropriate specialists.57,58 With regard to mental health, the prevalence of depression and anxiety may be increased among patients with CMT, especially in those with more severe forms.59,60 The available evidence is insufficient to establish specific recommendations on the pharmacological and non-pharmacological treatment of mental health disorders in patients with CMT.61 Regarding pregnancy, the evidence suggests a slight increase in the frequency of certain obstetric complications (eg, placenta praevia, abnormal fetal position, pre-term birth).62 Furthermore, approximately one-third of patients report worsening of neuropathic symptoms during pregnancy, although these improve after delivery in half of cases.63,64 Close neurological, gynaecological, and obstetric follow-up of pregnant women with CMT is therefore recommended.
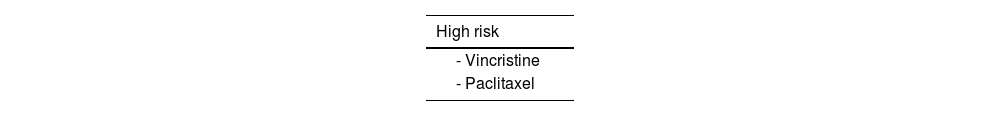
There is no evidence that patients with CMT are more vulnerable to neurotoxic drugs, except for vincristine and possibly paclitaxel, which may induce an atypical and more severe course of peripheral neurotoxicity.65 It is therefore essential that patients with CMT are not denied effective treatments that can prolong life expectancy in patients with cancer or improve health status in patients with non-oncological diseases. In any case, it is advisable to inform patients about potentially neurotoxic drugs that may worsen their symptoms and to include this information in clinical reports so that the patient can inform other healthcare professionals (Table 4). We also lack clear evidence demonstrating an increased risk associated with local anaesthesia or nerve blocks.66 Lastly, it should be noted that nitrous oxide can cause irreversible inactivation of cyanocobalamin; this drug should therefore be used with caution, after determination of vitamin B12 levels.65
Neurotoxic pharmacological treatments for Charcot-Marie-Tooth disease.
| High risk |
|---|
| - Vincristine |
| - Paclitaxel |
| Significant risk |
|---|
| - Adalimumab |
| - Amiodarone |
| - Nucleoside analogues |
| - Bevacizumab |
| - Bortezomib |
| - Ciclosporin A |
| - Cisplatin, carboplatin, oxaliplatin |
| - Chloramphenicol |
| - Chloroquine |
| - Colchicine |
| - Dapsone |
| - Disulfiram |
| - Statins |
| - Stavudine |
| - Ethambutol |
| - Etanercept |
| - Phenytoin |
| - Fluoroquinolones |
| - Hydralazine |
| - Infliximab |
| - Leflunomide |
| - Linezolid |
| - Metronidazole |
| - Nitrofurantoin |
| - Nitrous oxide (B12 deficiency) |
| - Pridoxine (high doses) |
| - Propafenone |
| - Sertraline |
| - Sorafenib |
| - Sulfasalazine |
| - Suramin |
| - Sunitinib |
| - Tacrolimus |
| - Thalidomide |
| - Taxoids |
Key concepts:
- •
Symptoms such as pain, fatigue, and cramps should be assessed periodically. The available evidence is insufficient to issue specific recommendations about the treatment of these symptoms in patients with CMT.
- •
Some patients may need other types of treatment, depending on their phenotypic characteristics (respiratory failure, optic neuropathy, etc) or associated comorbidities (depression, anxiety, SAHS, etc).
- •
Close neurological, gynaecological, and obstetric follow-up is recommended for pregnant women with CMT.
- •
Caution should be exercised, and patients should be informed about potentially neurotoxic drugs that may worsen their symptoms.
Rehabilitation therapy is an essential pillar of the treatment of CMT, and should be delivered by a multidisciplinary team including a medical rehabilitation specialist, a physiotherapist, an occupational therapist, and an orthotic/prosthetic technician to satisfy all functional needs. Early indication of rehabilitation therapy is key to identifying the patient’s functional deficits and establishing treatment objectives, which will vary over the course of follow-up. This enables the design of an individualised plan including therapeutic exercise, orthotic aids, and/or other technical support measures.67 Gait disfunction in patients with CMT results from alterations at different levels, which must be managed with the appropriate therapeutic approach:
- -
Joint deformities. Rehabilitation therapy is mainly based on orthoprosthetic adaptations to improve ankle stability and support. Evidence on each potential adaptation is limited, and the process should be guided by a multidisciplinary team. Various options can be used, such as insoles that unload areas of excess pressure or orthopaedic footwear with plaster casting aimed at maintaining the hindfoot in a neutral position.67,68 In children, a consensus statement recommends serial casting to maintain or improve ankle dorsiflexion, although use of this technique is not widespread.69 Furthermore, physical therapy techniques involving kinesiotherapy (passive or active-assisted exercises) and stretching of retracted or contracted muscles and the plantar fascia may be useful. The use of botulinum toxin in flexible pes cavus may also be considered.68
- -
Muscle weakness. The degree of muscle weakness must be assessed with the Medical Research Council (MRC) scale and/or dynamometry. The rehabilitation approach involves therapeutic physical exercise and orthoprosthetic adaptation, with a primary objective of compensating for the limitation in ankle dorsiflexion. Regarding orthoprosthetic adaptation in the initial stages, evidence supports the use of devices for equinus correction featuring an elastic band that lifts the forefoot.67,70 As weakness progresses, the most recommended orthotic device is the ankle-foot orthosis (AFO), typically with a double-action ankle joint and plantar flexion stop (PLS-AFO), which may be adapted according to the patient’s strength and range of motion.67 Several studies have shown the benefits of therapeutic physical exercise, although there is no consensus regarding the type, frequency, or intensity of exercise.71 In general terms, aerobic training programmes improve strength in adults, whereas in children, progressive resistance training programmes for foot dorsiflexion weakness have been found to improve strength and delay the progression of weakness.67,72,73
- -
Somatosensory deficits. Balance retraining programmes and proprioceptive exercises are recommended, although there is no evidence of their effectiveness for slowing disease progression. Furthermore, AFOs that block plantar flexion may improve sensory feedback, whereas walking aids improve gait stability.67
Fine motor skills may be impaired in patients with CMT, as a result of progressive weakness of the intrinsic muscles of the hand, resulting in reduced grip strength and reduced dexerity. In this context, an individualised assessment by an occupational therapist is essential in prescribing technical support measures that can improve patient function (eg, adapted cutlery, buttoning and zipping aids, etc). Thumb opposition splints can increase functionality in daily activities and improve occupational performance.74
Key concepts:
- •
Rehabilitation therapy should be initiated early to identify and improve functional deficits.
- •
It is important to assess the impact of joint deformities, muscle weakness, and somatosensory deficits on gait and fine motor skills.
- •
Specific, tailored therapeutic goals must be established, and modified as the disease progresses.
- •
Despite the lack of scientific evidence, the treating physicians should consider aerobic or muscle resistance exercise programmes, physical therapy, orthoprosthetic adaptations, and/or technical support measures.
Orthopaedic surgery for CMT focuses primarily on cavovarus deformity, although it may also be prescribed for the treatment of other less frequent deformities, such as scoliosis, arthrogryposis, or claw hand. There is no consensus in the literature regarding the exact timing for the referral of these patients to an orthopaedic surgeon. However, we recommend that the multidisciplinary team include an orthopaedic surgeon from the early stages. In any case, the decision to indicate surgery should be made on an individual basis, and patients should be duly informed of the short- and long-term risks and benefits of this treatment option.
The use of orthotic devices is often the initial treatment for joint deformities in patients with CMT. However, when the deformity becomes structural, surgical correction can achieve better outcomes. Deformities in the early stages can be corrected with osteotomy, tendon transfer, and fasciotomy or tenotomy; these surgical techniques help to preserve joints and improve control over the progression of deformities. However, it is important to inform patients that CMT is a dynamic entity in which new deformities may appear, potentially requiring corrective surgery.10,75
From a biomechanical viewpoint, patients with CMT usually present cavus, varus, and equinus deformities. Therefore, posterior tibial tendon transfer to the dorsum of the foot is a highly effective surgical approach, as it removes the deforming force and helps to increase dorsiflexion strength, which is often decreased in these patients due to the involvement of the anterior tibial tendon. Researchers agree that lengthening or partial tenotomy of the posterior tibial tendon is not indicated in the early stages, as it may result in a loss of strength.76,77 Regarding posterior tibial tendon transfer, it should be noted that dorsiflexion strength of the tendon is greater when the tendon is inserted directly onto the bone, and when it is inserted at more distal locations. Furthermore, maximal tension should not be applied when inserting the tendon on the dorsum of the foot. After the procedure, if the hindfoot remains in varus, the best option is correction with a calcaneal osteotomy.78 Likewise, there is consensus that transferring the long peroneal tendon to the short peroneal tendon helps to reduce the drop of the first metatarsal while increasing forefoot abduction strength. Finally, once the long peroneal tendon is detached, the surgeon must assess whether the drop of the first metatarsal persists. If that is the case, osteotomy should be considered to correct forefoot cavus and elevate the metatarsal.79
Other extra-articular surgical techniques include the sectioning of the plantar fascia and Achilles tenotomy. If the patient presents claw toe (including the hallux), the best option is arthrodesis combined with proximal transfer of the extensors, thus maintaining extension strength and preventing the persistence of the deformity.80 Finally, as a last resort, hindfoot and midfoot arthrodesis may be considered for patients presenting deterioration of the joint surface or stiffness preventing extra-articular correction. This type of intervention should be avoided to the greatest extent possible, especially in the early stages of the disease.
Key concepts:
- •
The multidisciplinary working group should include an orthopaedic surgeon from the early stages.
- •
Decision-making regarding surgical treatment should be individualised and informed.
- •
Flexible cavovarus foot can be treated surgically by transferring the tendon of the posterior tibial muscle to the dorsum of the foot. This procedure may be combined with calcaneal osteotomy or transfer of the long peroneal tendon to the short peroneal tendon.
- •
Fixation or arthrodesis should be avoided to the greatest extent possible in the early stages of the disease.
At the time of drafting these guidelines, no disease-modifying treatment has been proven to be effective in clinical trials. Therefore, patients should be invited to participate in clinical trials whenever possible. Most clinical trials conducted to date have focused on CMT1A, and aim to reduce the expression of the PMP22 gene. In this regard, several strategies (eg, progesterone receptor antagonists, RNA interference) have been successfully evaluated in animal models. However, attempts to translate these results to humans have so far been discouraging.81,82 One possible reason is the slowly progressive course of the disease, which hinders assessment of the results and comparison against a control group.83 A recent phase III trial (ClinicalTrials.gov identifier NCT03023540) explored a combination of baclofen, d-sorbitol, and naltrexone (PXT3003), with promising results, although the trial is currently under re-evaluation.84 In any case, the most promising treatments currently under development are those that focus on the underlying genetic defect.85
In other subtypes of CMT, several lines of research and animal models have explored the potential of different pharmacological treatments. Particularly interesting is the case of CMT/dHMN associated with pathogenic SORD variants, which has been found to be linked to increased intracellular sorbitol levels in fibroblasts. Aldose reductase inhibitors have been observed to lower intracellular sorbitol levels in fibroblasts and to improve motor symptoms in a Drosophila model.32
An attractive alternative to treatments targeting each genetic subtype is the search for pharmacological agents that act on pathogenic pathways common to several CMT subtypes. These agents include molecules promoting axonal transport, such as histone deacetylase 6 inhibitors, which have been shown to improve α-tubulin acetylation, motor performance, and electrophysiological parameters in a mouse model of CMT2-HSPB1 or in cell models of CMT2D caused by pathogenic GARS1 variants.86,87 Despite the promising results in cell and animal models, human studies are yet to be performed.
Key concepts:
- •
At the time of drafting these guidelines, no disease-modifying treatment has been shown to be efficacious in clinical trials.
- •
Patients should be offered the possibility to participate in clinical trials whenever possible.
The slowly progressive course of most subtypes of CMT, along with significant intra- and interfamilial variability, makes it difficult to reliably measure and predict progression, and to assess the efficacy of potential treatments.83 Follow-up assessments should be conducted at least once per year, and should include a neurological examination, with assessment of muscle strength by muscle groups using the MRC scale. Furthermore, evidence supports the use of simple clinical scales, such as the Charcot-Marie-Tooth Neuropathy Score (CMTNS) and its subscore excluding electrophysiology (Charcot-Marie-Tooth Examination Score [CMTES]). These tools allow for objective follow-up within and between centres, and help to better establish the stage of the disease.88,89 Many other clinical scales have been developed that may be useful for patient follow-up (Supplementary material 3).88–96 However, we do not consider their routine use necessary outside of clinical trials or research settings.
Periodic electrophysiological follow-up studies are not recommended, except in specific situations, such as unexpected clinical worsening. Routine performance of imaging studies is also not recommended for follow-up, although they may be useful in assessing disease progression.
Key concepts:
- •
Follow-up visits with patients with CMT should be held at least annually, by a multidisciplinary team specifically formed to meet the needs of each individual patient.
- •
Clinical evaluation should include simple sclaes such as the CMTES and the MRC scale.
- •
We do not recommend systematic, periodic electrophysiological studies.
This collaborative project, developed by a multidisciplinary expert group, provides a series of consensus recommendations on the diagnosis and management of patients with CMT. We present a series of diagnostic keys, as well as practical recommendations on neurophysiological, genetic, and other studies that provide diagnostic certainty. We also make recommendations for informing the patient of the diagnosis and providing genetic counselling, as well as for the follow-up and symptomatic, rehabilitation, and orthopaedic treatment of CMT.
Conflicts of interestThe authors have no conflicts of interest to declare.
The following are Supplementary data to this article:
