



Autosomal recessive spinocerebellar ataxia refers to a large group of diseases affecting the cerebellum and/or its connections, although they may also involve other regions of the nervous system. These diseases are accompanied by a wide range of systemic manifestations (cardiopathies, endocrinopathies, skeletal deformities, and skin abnormalities).
DevelopmentThis study reviews current knowledge of the most common forms of autosomal recessive spinocerebellar ataxia in order to provide tips that may facilitate diagnosis.
ConclusionsA thorough assessment of clinical phenotype (pure cerebellar or cerebellar-plus syndrome, with or without systemic manifestations), laboratory tests (vitamin E, acanthocytosis, albumin, cholesterol, phytanic acid, lactic acid, creatine kinase, cholestanol, coenzyme Q10, alpha-fetoprotein, copper, ceruloplasmin, chitotriosidase), nerve conduction studies (presence and type of neuropathy), and an magnetic resonance imaging study (presence of cerebellar atrophy, presence and location of signal alterations) may help establish a suspected diagnosis, which should be confirmed by detecting the underlying genetic mutation. A positive genetic test result is necessary to determine prognosis and provide adequate genetic counselling, and will also permit appropriate treatment of some entities (abetalipoproteinaemia, ataxia with vitamin E deficiency, Refsum disease, cerebrotendinous xanthomatosis, Niemann-Pick disease type C, Wilson disease). Without a genetic diagnosis, conducting basic research and therapeutic trials will not be possible.
Las ataxias espinocerebelosas de herencia recesiva constituyen un amplio grupo de enfermedades del cerebelo y/o de sus conexiones; en muchos casos también se afectan otras partes del sistema nervioso. Asimismo, y con cierta frecuencia, se acompañan de diversas manifestaciones sistémicas (cardiopatía, alteraciones cutáneas, endocrinopatías, malformaciones esqueléticas).
DesarrolloEn este trabajo se revisan los conocimientos actuales sobre las principales ataxias recesivas de curso progresivo, con el fin de establecer unas claves que faciliten su complejo diagnóstico.
ConclusionesUna cuidadosa evaluación clínica (síndrome espinocerebeloso puro o plus con o sin manifestaciones sistémicas), acompañada de la determinación de ciertos marcadores de laboratorio (vitamina E, acantocitosis, alfa-fetoproteína, ácido láctico, albúmina, colesterol, ácido fitánico, coenzima-Q10, CK, colestanol, quitotriosidasa, cobre, ceruloplasmina), la valoración de los datos del estudio electroneuromiográfico (ausencia o presencia de neuropatía y tipo de la misma) y los hallazgos del estudio de resonancia magnética (ausencia o presencia de atrofia cerebelosa, ausencia o presencia de alteraciones de la intensidad de señal y localizaciones de las mismas), ayudarán al clínico a establecer unas determinadas sospechas diagnósticas, que siempre procurará confirmar con la detección de la mutación genética causal. El hallazgo de la mutación es decisivo para establecer el pronóstico y consejo genético, además permitirá indicar un tratamiento eficaz en determinadas entidades (abetalipoproteinemia, ataxia por déficit de vitamina E, enfermedad de Refsum, xantomatosis cerebrotendinosa, enfermedad de Niemann-Pick tipo C, enfermedad de Wilson). Sin diagnóstico genético no será posible realizar investigación básica ni tampoco poner en marcha ensayos terapéuticos.
The 2 main definitions of ataxia (meaning “without order”) are balance disorders and a lack of motor coordination. The term was first used in 1864 by Duchenne de Boulogne,1 who described the gait disorder caused by tabes dorsalis as “locomotor ataxia.” A year previously, Nikolaus Friedreich2 had described the disease that now bears his name, the most frequent form of autosomal recessive ataxia. In the late 19th century, Pierre Marie distinguished Friedreich ataxia (FA) from dominant forms of hereditary ataxia.
Degenerative ataxias progressively alter motor function due to involvement of the cerebellum and/or its main connections (the cerebral cortex, posterior columns, spinocerebellar tracts, vestibular nuclei, red nuclei, pontine nuclei, and the inferior olivary nuclei).3 In addition to imbalance (midline ataxia), poor limb coordination (dysmetria/dysdiadochokinesia), dysarthria, oscillopsia, and nystagmus, patients with ataxia may also display intellectual disability or cognitive impairment, epilepsy, pyramidal signs, abnormal movement, visual alterations, hearing loss, lower motor neuron disease, peripheral neuropathy, and myopathy. The cerebellum also has a role in executive function and emotional and language processing, which can be affected in hereditary ataxias.4–6
The aim of this article, based on a literature search on the PubMed, OMIM, and GeneReviews databases, in addition to information and images from the author's own patients, is to establish a systematic diagnostic protocol for degenerative, progressive, autosomal recessive ataxias (excluding such congenital ataxias as Joubert syndrome, Norman syndrome, Gillespie syndrome, Pollitt syndrome, etc.). Particular emphasis is placed on the most frequent and treatable forms.
DevelopmentPatients with ataxiaClinical history can be used to profile the onset and progression of the condition (progressive, non-progressive, or episodic ataxia), the type of spinocerebellar syndrome (midline, appendicular, or mixed), and the possible involvement of other systems (cerebellar-plus or pure cerebellar syndromes). Other useful information can be obtained from the patient's personal history (toxic agents, medication, deficiency diseases, endocrinopathies, infections, tumours, autoimmune diseases, prionopathies)7 and family history (inheritance pattern: dominant, recessive, X-linked, or maternal mitochondrial). The term spinocerebellar ataxia is used to describe progressive ataxias following an autosomal dominant inheritance pattern, whereas recessive ataxias characterised mainly by cerebellar atrophy are referred to as autosomal recessive cerebellar ataxias (ARCA). The Scale for the Assessment and Rating of Ataxia (SARA)8 and the International Cooperative Ataxia Rating Scale (ICARS)9 are useful for monitoring progression, while the EQ-5D and other quality of life scales are used to assess the health effects of the disease.
Misdiagnosis or delayed diagnosis of ataxia can be caused by a range of factors: patients denying or minimising their symptoms, instability being attributed to emotional disorders or stress, patients being referred to other specialists (traumatologists, rheumatologists, ophthalmologists, otorhinolaryngologists, etc.), or re-assessment not being performed following initial consultation of a new case.10
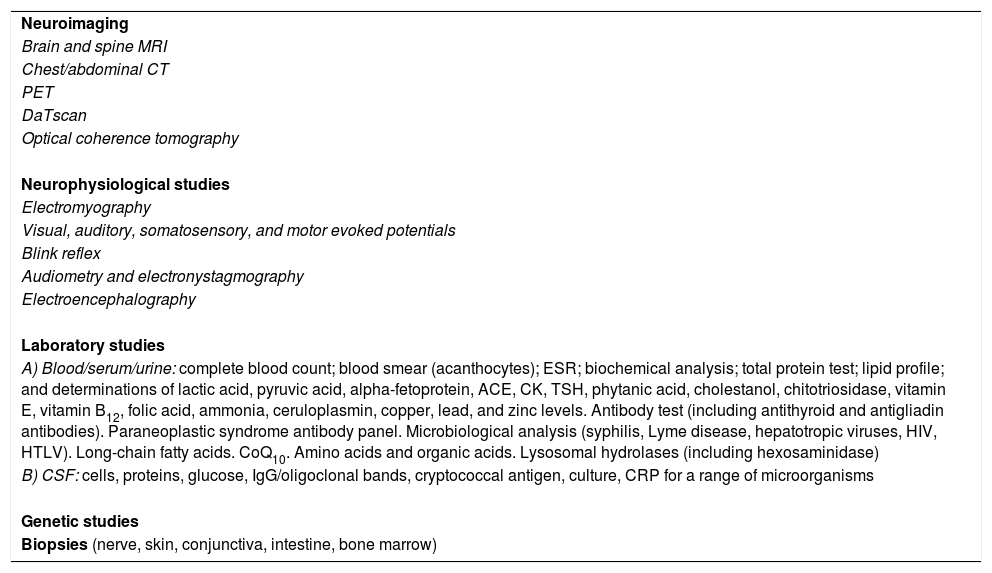
Depending on the diagnostic hypothesis proposed following the clinical history interview, these patients may need to undergo extensive testing. Table 1 gives details on the entire battery of complementary tests, which should be used selectively according to the case.
Diagnostic tests that may be necessary in patients with progressive ataxia of unknown origin.
| Neuroimaging |
| Brain and spine MRI |
| Chest/abdominal CT |
| PET |
| DaTscan |
| Optical coherence tomography |
| Neurophysiological studies |
| Electromyography |
| Visual, auditory, somatosensory, and motor evoked potentials |
| Blink reflex |
| Audiometry and electronystagmography |
| Electroencephalography |
| Laboratory studies |
| A) Blood/serum/urine: complete blood count; blood smear (acanthocytes); ESR; biochemical analysis; total protein test; lipid profile; and determinations of lactic acid, pyruvic acid, alpha-fetoprotein, ACE, CK, TSH, phytanic acid, cholestanol, chitotriosidase, vitamin E, vitamin B12, folic acid, ammonia, ceruloplasmin, copper, lead, and zinc levels. Antibody test (including antithyroid and antigliadin antibodies). Paraneoplastic syndrome antibody panel. Microbiological analysis (syphilis, Lyme disease, hepatotropic viruses, HIV, HTLV). Long-chain fatty acids. CoQ10. Amino acids and organic acids. Lysosomal hydrolases (including hexosaminidase) |
| B) CSF: cells, proteins, glucose, IgG/oligoclonal bands, cryptococcal antigen, culture, CRP for a range of microorganisms |
| Genetic studies |
| Biopsies (nerve, skin, conjunctiva, intestine, bone marrow) |
ACE: angiotensin-converting enzyme; CK: creatine kinase; CoQ10: coenzyme Q10; CRP: C-reactive protein; CSF: cerebrospinal fluid; CT: computed tomography; ESR: erythrocyte sedimentation rate; HIV: human immunodeficiency virus; HTLV: human T-cell lymphotropic virus; IgG: inmunoglobulin G; MRI: magnetic resonance imaging; PET: positron emission tomography; TSH: thyroid stimulating hormone.
Symptoms of recessive ataxias usually appear in the first 3 decades of life, although cases of neonatal and delayed onset have also been reported; progression is often faster than in dominant forms. Pure cerebellar syndrome does occur, although it is more common for patients also to present symptoms secondary to the involvement of other systems, such as intellectual disability, cognitive impairment, epilepsy, movement disorders (dystonia, chorea, parkinsonism, tremor), pyramidal signs, oculomotor dysfunction (oculomotor apraxia, supranuclear vertical gaze palsy), peripheral neuropathy, retinal dysfunction, or hearing loss. Other organs may also be involved (cardiomyopathy, diabetes, hepatosplenomegaly, musculoskeletal disorders).10–15
FA is the most common form of recessive ataxia, followed by ataxia-telangiectasia (AT), ataxia with oculomotor apraxia type 1 (AOA1), and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS).15,16 Some types of progressive ataxia are associated with biomarkers. Neuroimaging studies, particularly magnetic resonance imaging (MRI), may provide key information for diagnosing some recessive ataxias; these are described in the specific sections for each form of ataxia. Electrophysiological study of peripheral nerves is of great help in confirming or ruling out the presence or type of neuropathy (motor, sensory, axonal, demyelinating, mixed).
Treatments are available that can modify the course of a group of ataxias caused by metabolic dysfunction (abetalipoproteinaemia [ABLP], ataxia with vitamin E deficiency [AVED], Refsum disease, cerebrotendinous xanthomatosis [CX], Niemann-Pick disease type C, Wilson disease, and ARCA2)17; this type of ataxia should always be considered in differential diagnosis.10 Other metabolic disorders, such as biotinidase deficiency, urea cycle disorders, Hartnup disease, alpha-methylacyl-CoA racemase deficiency, and gamma-glutamylcysteine synthetase deficiency, can also manifest with multisystemic symptoms, including early-onset ataxia.
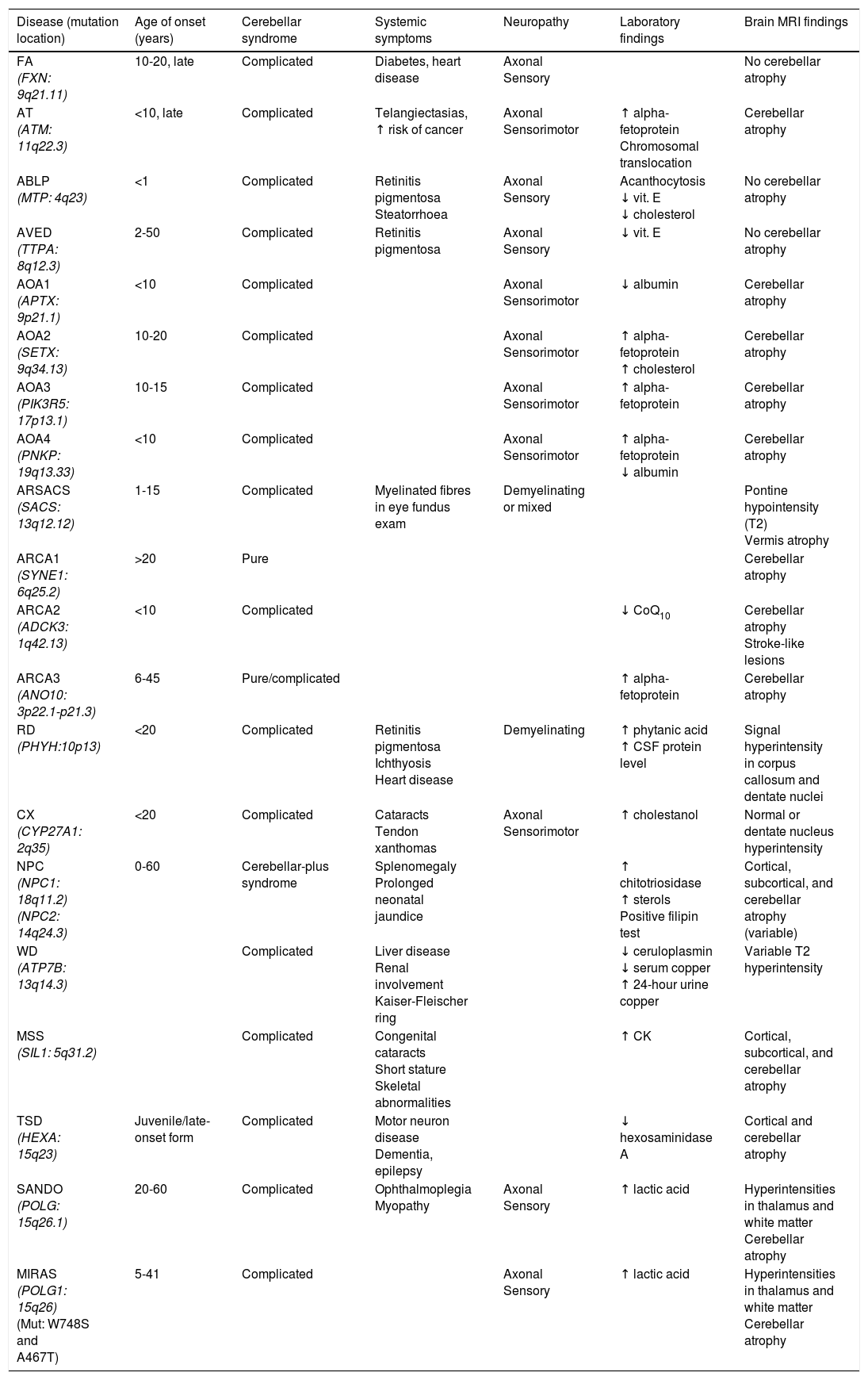
Table 2 includes a summary of the clinical, electromyographic, laboratory, and MRI findings for the most frequent autosomal recessive ataxias.
Main recessive disorders causing progressive ataxia.
| Disease (mutation location) | Age of onset (years) | Cerebellar syndrome | Systemic symptoms | Neuropathy | Laboratory findings | Brain MRI findings |
|---|---|---|---|---|---|---|
| FA (FXN: 9q21.11) | 10-20, late | Complicated | Diabetes, heart disease | Axonal Sensory | No cerebellar atrophy | |
| AT (ATM: 11q22.3) | <10, late | Complicated | Telangiectasias, ↑ risk of cancer | Axonal Sensorimotor | ↑ alpha-fetoprotein Chromosomal translocation | Cerebellar atrophy |
| ABLP (MTP: 4q23) | <1 | Complicated | Retinitis pigmentosa Steatorrhoea | Axonal Sensory | Acanthocytosis ↓ vit. E ↓ cholesterol | No cerebellar atrophy |
| AVED (TTPA: 8q12.3) | 2-50 | Complicated | Retinitis pigmentosa | Axonal Sensory | ↓ vit. E | No cerebellar atrophy |
| AOA1 (APTX: 9p21.1) | <10 | Complicated | Axonal Sensorimotor | ↓ albumin | Cerebellar atrophy | |
| AOA2 (SETX: 9q34.13) | 10-20 | Complicated | Axonal Sensorimotor | ↑ alpha-fetoprotein ↑ cholesterol | Cerebellar atrophy | |
| AOA3 (PIK3R5: 17p13.1) | 10-15 | Complicated | Axonal Sensorimotor | ↑ alpha-fetoprotein | Cerebellar atrophy | |
| AOA4 (PNKP: 19q13.33) | <10 | Complicated | Axonal Sensorimotor | ↑ alpha-fetoprotein ↓ albumin | Cerebellar atrophy | |
| ARSACS (SACS: 13q12.12) | 1-15 | Complicated | Myelinated fibres in eye fundus exam | Demyelinating or mixed | Pontine hypointensity (T2) Vermis atrophy | |
| ARCA1 (SYNE1: 6q25.2) | >20 | Pure | Cerebellar atrophy | |||
| ARCA2 (ADCK3: 1q42.13) | <10 | Complicated | ↓ CoQ10 | Cerebellar atrophy Stroke-like lesions | ||
| ARCA3 (ANO10: 3p22.1-p21.3) | 6-45 | Pure/complicated | ↑ alpha-fetoprotein | Cerebellar atrophy | ||
| RD (PHYH:10p13) | <20 | Complicated | Retinitis pigmentosa Ichthyosis Heart disease | Demyelinating | ↑ phytanic acid ↑ CSF protein level | Signal hyperintensity in corpus callosum and dentate nuclei |
| CX (CYP27A1: 2q35) | <20 | Complicated | Cataracts Tendon xanthomas | Axonal Sensorimotor | ↑ cholestanol | Normal or dentate nucleus hyperintensity |
| NPC (NPC1: 18q11.2) (NPC2: 14q24.3) | 0-60 | Cerebellar-plus syndrome | Splenomegaly Prolonged neonatal jaundice | ↑ chitotriosidase ↑ sterols Positive filipin test | Cortical, subcortical, and cerebellar atrophy (variable) | |
| WD (ATP7B: 13q14.3) | Complicated | Liver disease Renal involvement Kaiser-Fleischer ring | ↓ ceruloplasmin ↓ serum copper ↑ 24-hour urine copper | Variable T2 hyperintensity | ||
| MSS (SIL1: 5q31.2) | Complicated | Congenital cataracts Short stature Skeletal abnormalities | ↑ CK | Cortical, subcortical, and cerebellar atrophy | ||
| TSD (HEXA: 15q23) | Juvenile/late-onset form | Complicated | Motor neuron disease Dementia, epilepsy | ↓ hexosaminidase A | Cortical and cerebellar atrophy | |
| SANDO (POLG: 15q26.1) | 20-60 | Complicated | Ophthalmoplegia Myopathy | Axonal Sensory | ↑ lactic acid | Hyperintensities in thalamus and white matter Cerebellar atrophy |
| MIRAS (POLG1: 15q26) (Mut: W748S and A467T) | 5-41 | Complicated | Axonal Sensory | ↑ lactic acid | Hyperintensities in thalamus and white matter Cerebellar atrophy |
ABLP: abetalipoproteinaemia; AOA: ataxia with oculomotor apraxia; ARCA: autosomal recessive cerebellar ataxia; ARSACS: autosomal recessive spastic ataxia of Charlevoix-Saguenay; AT: ataxia-telangiectasia; AVED: ataxia with vitamin E deficiency; CK: creatine kinase; CSF: cerebrospinal fluid; CX: cerebrotendinous xanthomatosis; FA: Friedreich ataxia; MIRAS: mitochondrial recessive ataxia syndrome; MSS: Marinesco-Sjögren syndrome; NPC: Niemann-Pick disease type C; RD: Refsum disease; SANDO: sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; TSD: Tay-Sachs disease; WD: Wilson disease.
Ophthalmological examination may be of great assistance when a specific type of autosomal recessive ataxia is suspected. Typical findings include telangiectasia of the sclera in AT, cataracts in CX and in Marinesco-Sjögren syndrome, Kayser-Fleischer ring in Wilson disease, myelinated fibres in eye fundus examination and increased thickness of the nerve fibre layer in optical coherence tomography imaging in ARSACS, and retinitis pigmentosa in ABLP and AVED. Ichthyosis is indicative of Refsum disease. More than half of patients with Niemann-Pick disease type C present splenomegaly; many have a history of prolonged neonatal jaundice. CX is so named due to the presence of tendon xanthomas, although these may be absent. The scoliosis-cardiopathy-diabetes triad of symptoms is part of the clinical spectrum of FA.
Brief description of the most significant autosomal recessive ataxiasFriedreich ataxiaFA is caused by mutations to the FXN gene (9q21.11), which codes for frataxin, a protein that plays a role in mitochondrial function by activating oxidative phosphorylation (absence of the protein causes accumulation of mitochondrial iron, leading to oxidative stress and cell death). The mutation most frequently observed is an intronic GAA trinucleotide repeat expansion in homozygosis18; 2% of patients carry a missense mutation in one allele and a trinucleotide repeat expansion in the other. FA is the most frequent autosomal recessive ataxia in white populations. Age of onset ranges from 5 to 25 years, although cases have been described of late (25-39 years) and very late onset (40 years and older).19–21 The typical phenotype is progressive spinal and cerebellar ataxia (loss of sensory ganglion cells and degeneration of the posterior columns and spinocerebellar tracts), with areflexia, pes cavus, Babinski sign, and scoliosis; within 10 to 15 years of onset, patients have to use wheelchairs.22 In addition to axonal sensory polyneuropathy, auditory and optic neuropathy are also frequently observed. Hypertrophic cardiomyopathy and diabetes are common presentations of the condition. In cases of late or very late onset, FA may present as ataxia associated with some degree of paraparesis with retained tendon reflexes, or with symptoms similar to those of multiple system atrophy21; MRI reveals cerebellar and brainstem atrophy. Treatment with idebenone (a derivative of coenzyme Q10 [CoQ10]) may slow the development of cardiomyopathy and the progression of neurological impairment.17
Ataxia-telangiectasiaAT is caused by mutations to the ATM gene (11q22.3),23 which codes for a protein kinase involved in DNA repair; the phenotypic variability of AT is due to changes to the quantity/function of the protein.24,25 Symptom onset usually occurs in early childhood, although cases have been observed of much later onset. AT usually presents as midline ataxia, progressing to pancerebellar ataxia, with marked cerebellar atrophy. AT is distinguished from FA by this atrophy, and by the axonal sensorimotor neuropathy observed in these patients. Telangiectasias of the skin and sclera are characteristic of the condition, although they may not appear initially. Oculomotor apraxia and choreoathetosis are frequent symptoms. Patients present varying degrees of immunodeficiency, and are at risk of developing cancer (leukaemia, lymphoma). High serum levels of carcinoembryonic antigen or alpha-fetoprotein are constant markers, although alpha-fetoprotein is also observed at elevated levels in AOA2, AOA3, AOA4, and ARCA3. These conditions are also associated with DNA repair alterations.26 The detection of 7;14 translocations in karyotyping or increased radiosensitivity in vitro supports the diagnosis of AT, which is currently based on the identification of ATM mutations. There is no specific treatment for AT.
Another group of conditions referred to as AT-like disorders includes AT-LD1, which is caused by mutations to the MRE11A gene (11q21) and presents with similar symptoms to AT, with increased radiosensitivity, but with no telangiectasias or immunodeficiency.27
Abetalipoproteinaemia and ataxia with vitamin E deficiencyABLP is caused by mutations to the MTP gene (4q23), which codes for the large subunit of the heterodimeric microsomal triglyceride transfer protein, which is responsible for apolipoprotein B (chylomicrons and very-low-density lipoproteins) assembly.28 Onset usually occurs during the first year of life. Symptoms are similar to those observed in patients with FA, but are also associated with a malabsorption syndrome, with hypocholesterolaemia and deficiencies of fat-soluble vitamins (including vitamin E), as well as acanthocytosis and retinitis pigmentosa. Symptoms improve with diet therapy and vitamin supplements.
AVED is caused by mutations to the TTPA gene (8q12.3), coding for alpha-tocopherol transfer protein, which is responsible for binding vitamin E to very-low-density lipoproteins in the liver for subsequent transport to the nervous system. The condition presents similarly to FA, although with the additional symptom of decreased visual acuity secondary to retinitis pigmentosa; cardiomyopathy and diabetes also tend to be absent in AVED. Patients may also display dystonia, titubation (tremor) of the head, psychiatric symptoms, or cognitive impairment.29,30 It is essential to determine vitamin E levels in cases of ataxia of unknown origin, as vitamin E supplementation halts the progression of AVED.31
A specific type of autosomal recessive ataxia, caused by mutations to the ATCAY gene (19p13.3), is observed on Grand Cayman Island; this form also affects vitamin E metabolism.
Ataxia with oculomotor apraxia types 1 to 4AOA1 is caused by mutations to the APTX gene (9p21.1), which codes for aprataxin, a protein involved in DNA repair.32 Ataxia usually appears before the age of 10 years and is often associated with neuropathy, choreoathetosis, nystagmus, and oculomotor apraxia. The clinical presentation bears similarities to that of AT; however, patients with AOA1 frequently display hypoalbuminaemia and hypercholesterolaemia, which are not observed in AT.33 Some patients respond to CoQ10.34
AOA2 is caused by mutations to the SETX gene (9q34.13), which codes for senataxin, a helicase involved in DNA transcription and repair and the processing of several types of RNA.35 The condition is clinically similar to AOA1, but with later onset (in the second decade of life), and patients do not present hypoalbuminaemia or hypercholesterolaemia; elevated levels of alpha-fetoprotein are observed,36,37 however, for which reason differential diagnosis must consider AT. SETX missense mutations in heterozygosis have been described as a cause of juvenile amyotrophic lateral sclerosis type 4.38
AOA3 is caused by mutations to the PIK3R5 gene (17p13.1), and has been described in a Saudi Arabian family.39 Symptom onset occurs in the second decade of life and is associated with axonal sensorimotor polyneuropathy. Patients display high levels of alpha-fetoprotein.
AOA4 is caused by mutations to the PNKP gene (19q13.33). The condition has recently been described in Portugal,40 where AOA1 is also relatively prevalent. Symptoms appear in the first decade of life and progress rapidly. The condition is associated with sensorimotor polyneuropathy and occasionally dementia. Increased alpha-fetoprotein levels have been reported in some cases.
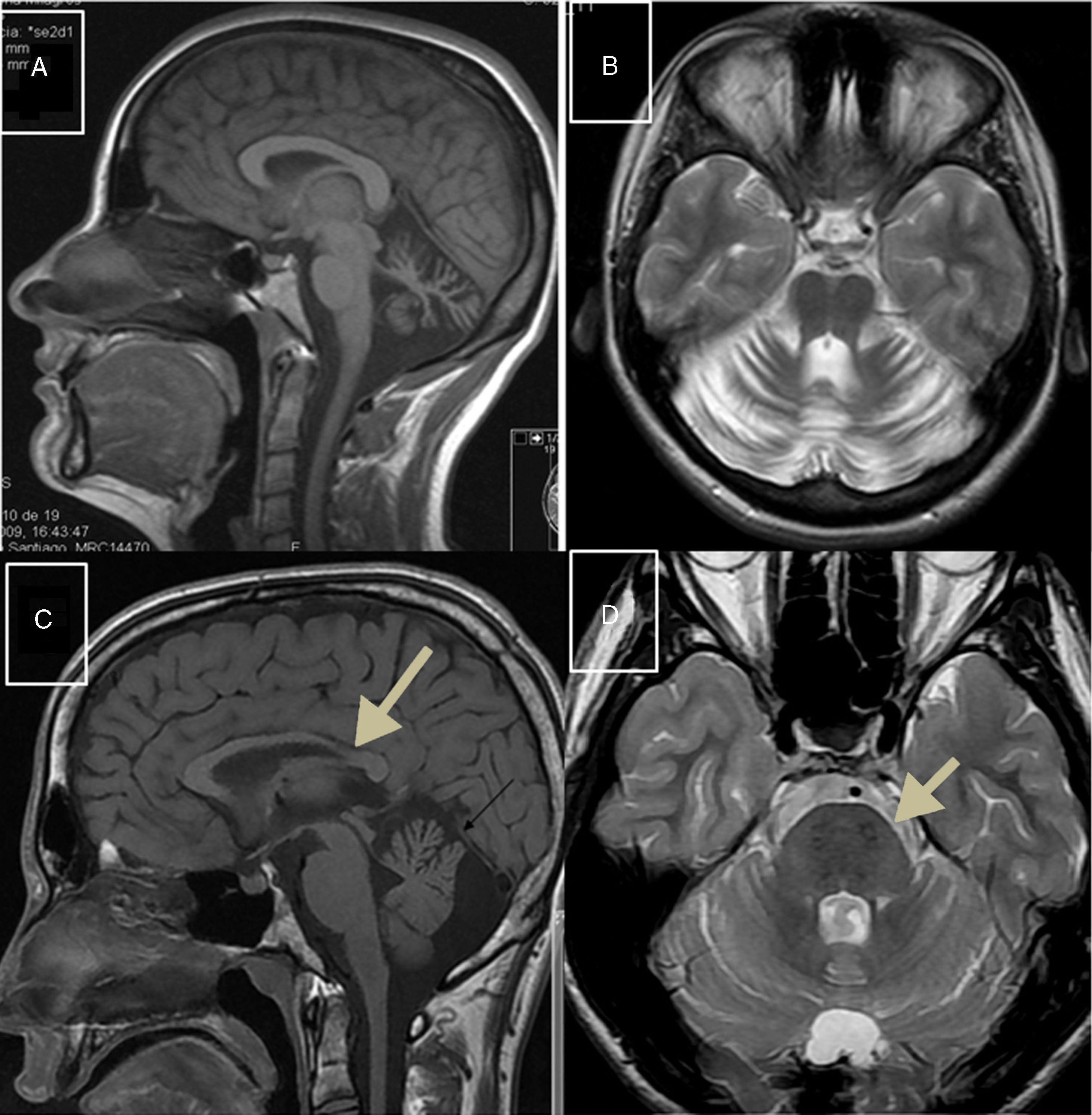
Autosomal recessive spastic ataxia of Charlevoix-SaguenayARSACS is caused by mutations to the SACS gene (13q12.12), which codes for sacsin, a chaperone protein that interacts with the proteasome and is involved in mitochondrial function and axonal and dendritic transport.41 The condition is thought to result from a developmental disorder and a degenerative process.42 It was first described in Quebec; cases have subsequently been reported in all regions of the world. The phenotype of the condition is relatively homogeneous and is characterised by early-onset ataxia accompanied by pyramidal signs, mixed sensorimotor neuropathy, and pes cavus. In some cases of late-onset ARSACS, pyramidal signs are less severe and patients display hearing loss. Examination of the eye fundus may reveal the persistence of myelinated fibres, whereas optical coherence tomography images may show increased thickness of the retinal nerve fibre layer. MRI studies show atrophy of the cerebellar vermis and the posterior part of the corpus callosum, and linear hypointensities in the pons (Fig. 1C and D); diffusion tensor imaging reveals alterations to the pontocerebellar fibres.42,43
 Sagittal T1-weighted sequence showing marked atrophy of the cerebellar vermis in a patient with ARCA1. B) Axial T2-weighted sequence showing pancerebellar atrophy in a patient with ARCA1. C) Axial T1-weighted sequence showing atrophy of the posterior part of the corpus callosum in a patient with ARSACS. D) Axial T2-weighted sequence showing linear hypointensities in the pons of a patient with ARSACS.")
MR images. A) Sagittal T1-weighted sequence showing marked atrophy of the cerebellar vermis in a patient with ARCA1. B) Axial T2-weighted sequence showing pancerebellar atrophy in a patient with ARCA1. C) Axial T1-weighted sequence showing atrophy of the posterior part of the corpus callosum in a patient with ARSACS. D) Axial T2-weighted sequence showing linear hypointensities in the pons of a patient with ARSACS.
ARCA1 is caused by mutations to the SYNE1 gene (6q25.2), which codes for nesprin-1, a nuclear membrane protein involved in linking the nucleoskeleton to the cytoskeleton. The condition is usually associated with a slowly progressive pure cerebellar syndrome, with MRI studies displaying evident pancerebellar atrophy (Fig. 1A and B). Onset usually occurs late in the third decade of life.44,45
ARCA2 is caused by mutations to the ADCK3 gene (1q42.13), resulting in primary CoQ10 deficiency. The condition is characterised by slowly progressive cerebellar ataxia, with onset in the first decade of life, sometimes associated with moderate intellectual disability, myoclonus, exercise intolerance, stroke-like episodes, and high blood lactic acid levels.46 MRI studies may display cortical hyperintensities in addition to the cerebellar atrophy.
ARCA3 is caused by mutations to the ANO10 gene (3p22.1-p21.3), and is characterised by ataxia with pyramidal signs and amyotrophy; age of onset ranges between 6 and 45 years.47 High serum alpha-fetoprotein levels have been reported in some patients.
Refsum diseaseRefsum disease is caused by mutations to the PHYH or PAHX gene (10p13),48 which codes for the peroxisomal enzyme phytanoyl-CoA hydroxylase. A deficiency of this protein causes an accumulation of phytanic acid, which damages myelin. Symptoms usually appear before the age of 20 years, and include ataxia, polyneuropathy with high CSF protein levels, hearing loss, retinitis pigmentosa, anosmia, epilepsy, and cognitive impairment in some cases; patients may also display ichthyosis, kidney and heart problems, and skeletal deformities.49 MRI studies reveal non-symmetrical hyperintensities in the corpus callosum, pyramidal tracts, and dentate nuclei. Lesions may show contrast uptake, which suggests active demyelination. Restriction of dietary phytanic acid, and plasmapheresis in cases of heart arrhythmia, improve disease progression.
Mutations to the PEX7 gene (6q23.3), which codes for the receptor peroxin-7, can cause a similar phenotype to that observed in Refsum disease, or more severe, complex conditions, such as rhizomelic chondrodysplasia punctata type 1, neonatal adrenoleukodystrophy, or Zellweger syndrome.50
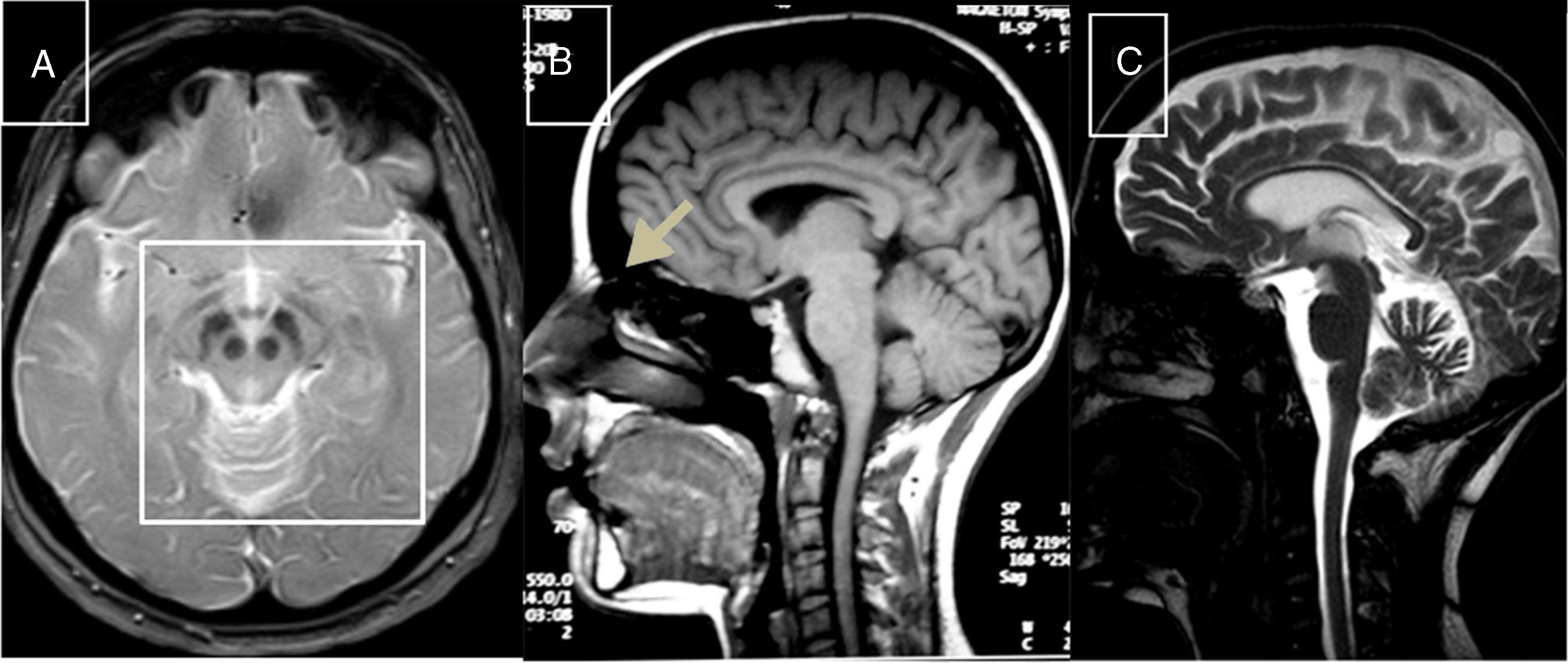
Cerebrotendinous xanthomatosisCX is caused by mutations to the CYP27A1 gene (2q35), which codes for sterol 27-hydroxylase, an enzyme involved in bile acid synthesis.51 Deficiency of this enzyme causes an accumulation of various sterols, including cholestanol, which is observed at high levels in the serum. Neurological symptoms usually appear late in the second decade of life, and comprise epilepsy, ataxia, pyramidal signs, and polyneuropathy. Psychiatric disorders are also common. Prior to onset of neurological symptoms, patients often have diarrhoea, cataracts, and tendon xanthomas; however, 3 patients from 2 different families have been observed not to display this sign, leading to diagnostic delays.52,53 MRI sometimes reveals hyperintensities in the dentate nuclei, enlarged cranial sinuses, and cervical spinal cord atrophy (Fig. 2B). Administration of chenodeoxycholic acid slows the progression of CX.
 Axial T2-weighted sequence from a patient with Wilson disease, showing periaqueductal hyperintensity, contrasting with hypointensity in the mammillary bodies, red nuclei, and substantia nigra, forming the “panda sign.” B) Sagittal T1-weighted sequence showing enlarged cranial sinuses and spinal cord atrophy in a patient with cerebrotendinous xanthomatosis. C) T2-weighted sequence showing atrophy in the cortex, corpus callosum, brainstem, and cerebellar vermis in a patient with Niemann-Pick disease type C.")
MR images of treatable hereditary ataxias secondary to metabolic disorders. A) Axial T2-weighted sequence from a patient with Wilson disease, showing periaqueductal hyperintensity, contrasting with hypointensity in the mammillary bodies, red nuclei, and substantia nigra, forming the “panda sign.” B) Sagittal T1-weighted sequence showing enlarged cranial sinuses and spinal cord atrophy in a patient with cerebrotendinous xanthomatosis. C) T2-weighted sequence showing atrophy in the cortex, corpus callosum, brainstem, and cerebellar vermis in a patient with Niemann-Pick disease type C.
This condition is caused by mutations to the NPC1 gene (18q11.2), coding for a transmembrane protein (90% of cases), or NPC2 (14q24.3), coding for an intralysosomal protein (10%). Deficiencies of these proteins distort the trafficking of cholesterol, sphingosine, and glycolipids, which accumulate in the lysosomes, leading to cell death.54 Clinical phenotypes vary according to age of onset: the neonatal form is fatal; the early childhood form causes hypotonia and psychomotor retardation; the late childhood form causes poor coordination, gait and language acquisition disorders, and gelastic cataplexy; the juvenile form causes ataxia, epilepsy, cataplexy, and poor school performance; and the adult form causes psychiatric disorders, ataxia, dystonia, and dementia.55 New massively parallel sequencing techniques have enabled more late-onset cases to be identified.56 Vertical gaze palsy is usually a symptom of the latter 3 forms. Patients often have a history of splenomegaly and prolonged neonatal jaundice. MRI scans reveal brain, cerebellar, and brainstem atrophy (Fig. 2C). Chitotriosidase in macrophages and CCL18 in the plasma do not always display high levels of activity. Filipin staining of fibroblast cultures reveals an accumulation of cholesterol. Administration of miglustat may improve disease progression.
Wilson diseaseWilson disease is caused by mutations to the ATP7B gene (13q14.3), which codes for an ATPase involved in transporting copper across the Golgi apparatus in hepatocytes.57 It can present in different forms, including fulminant hepatitis, psychosis, and neurological disorders (tremor, dystonia, akinetic-rigid syndrome, and occasionally ataxia). Most patients with neurological symptoms display the Kayser-Fleischer ring.58 MRI findings (Fig. 2A) vary greatly, as has previously been described.59 Laboratory testing shows low blood ceruloplasmin and copper levels and increased urine copper levels. The habitual treatment comprises D-penicillamine and trientine as chelants, as well as tetrathiomolybdate and zinc sulfate.59
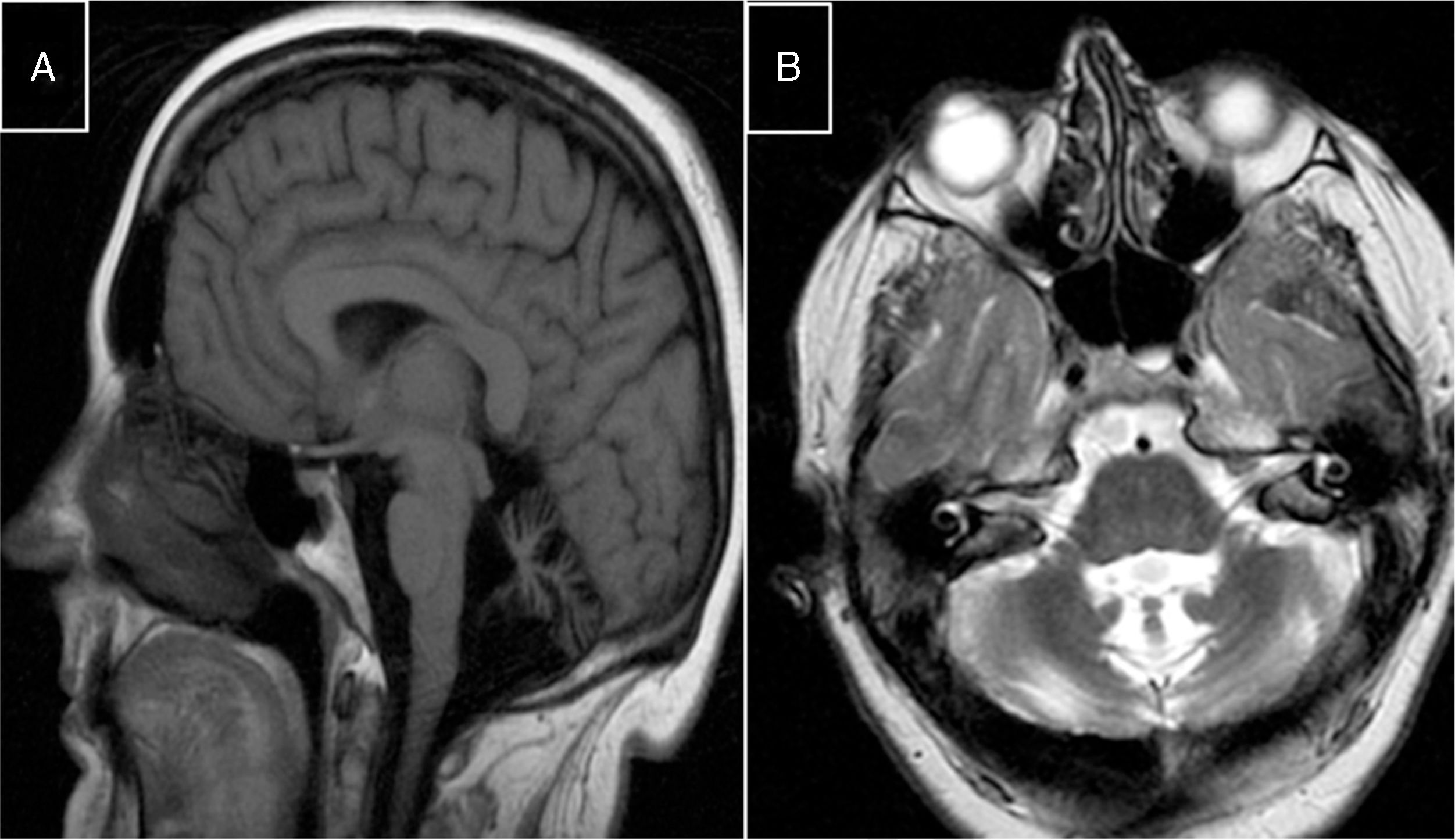
Marinesco-Sjögren syndromeThis condition is caused by mutations to the SIL1 gene (5q31.2), which codes for a nucleotide exchange factor for HSPA5, a heat-shock protein 70 (HSP-70) chaperone.60 Symptoms include short stature, hypergonadotropic hypogonadism, skeletal deformities, myopathy, congenital cataracts, and cerebellar ataxia; MRI studies display marked atrophy (Fig. 3).
 Sagittal T1-weighted sequence showing atrophy of the brainstem and cerebellar vermis. B) Axial T2-weighted sequence showing atrophy of the pons, vermis, and both cerebellar hemispheres, with no intraparenchymal signal alterations.")
MR images from a patient with Marinesco-Sjögren syndrome. A) Sagittal T1-weighted sequence showing atrophy of the brainstem and cerebellar vermis. B) Axial T2-weighted sequence showing atrophy of the pons, vermis, and both cerebellar hemispheres, with no intraparenchymal signal alterations.
Tay-Sachs disease is caused by dysfunction of the enzyme beta-hexosaminidase, encoded by the HEXA gene (15q23), and primarily affects the Ashkenazi Jewish population.61 Onset can take several forms: the infantile form causes hypotonia, loss of sight (cherry-red marks on the fundus), severe intellectual disability, and death at the age of approximately 3 years, and juvenile and adult forms can cause ataxia, lower motor neuron disease with atrophy and fasciculations, spasticity, dementia, and epilepsy.62
Mitochondrial recessive ataxia syndromeMutations to the POLG(1) gene (15q26.1), which codes for a subunit of a mitochondrial DNA polymerase, can result in a range of clinical presentations with different inheritance patterns. These include an ataxia syndrome with onset in children or young adults and associated with sensory neuropathy, dysarthria, and ophthalmoplegia. In some cases symptoms also include lactic acidosis, epilepsy, myopathy with ragged red fibres, and liver disease. MR images display thalamic hyperintensities and cerebellar atrophy.63 These disorders may follow either a dominant or a recessive inheritance pattern and can cause secondary mitochondrial DNA deletions. Mitochondrial recessive ataxia syndrome is caused by the A467T and W748S mutations in POLG and is a frequent cause of ataxia in Finland, Norway, and some Central European countries.64 Ataxia can also be caused by mutations to other genes related to mitochondrial function, such as RRM2B (8q22.3).65
Spinocerebellar ataxia with onset during childhood is caused by homo- or heterozygous mutations to the C10orf2 gene (10q23.3-24.1), which codes for the twinkle and twinky proteins. These mutations cause mitochondrial DNA depletion syndrome 7, which may present with ophthalmoplegia, hearing loss, early-onset spinocerebellar ataxia, and refractory epilepsy.66 This syndrome is the second-largest cause of ataxia in Finland.
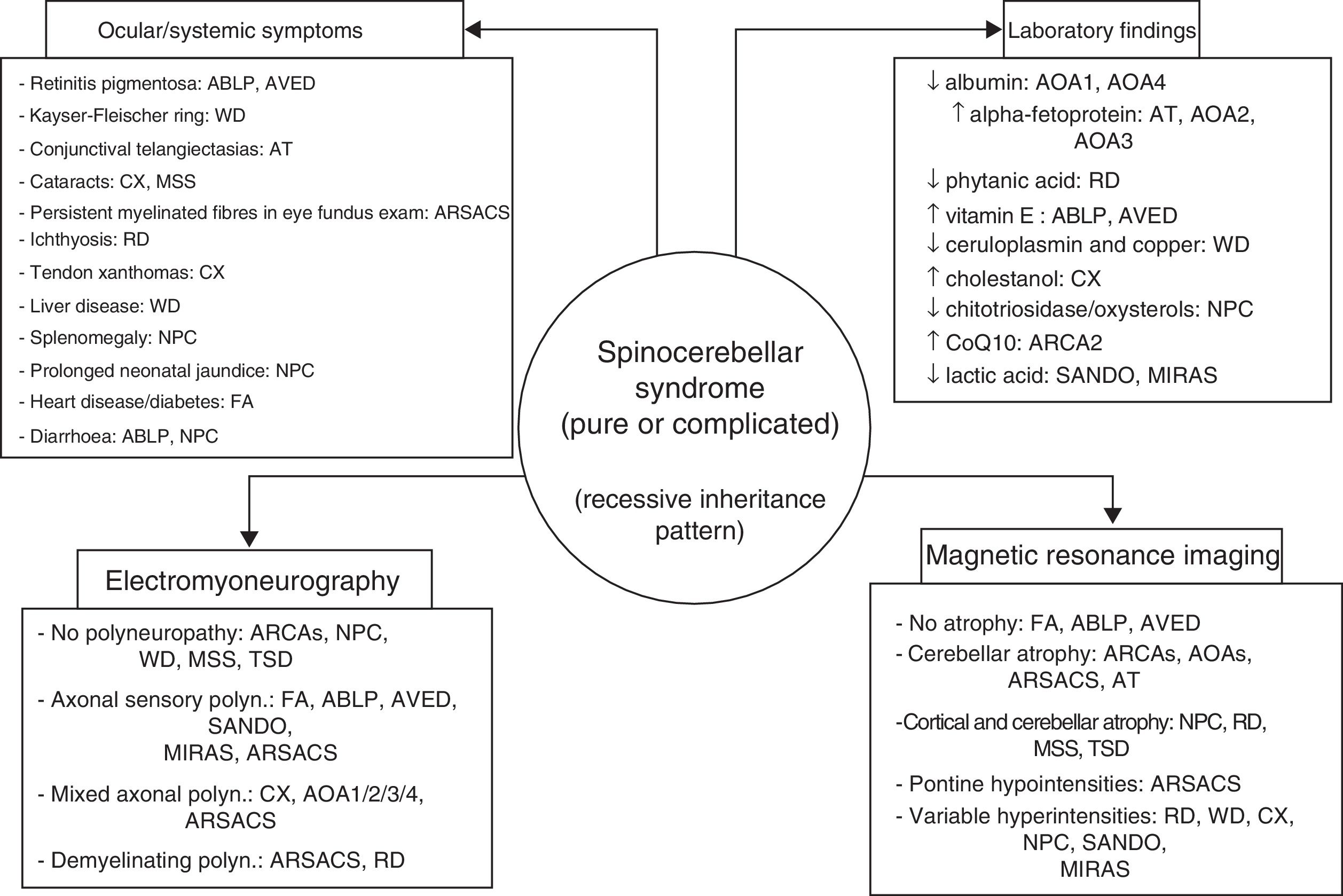
ConclusionsAutosomal recessive ataxias can present with a wide range of symptoms; different forms often overlap, and clinicians may have the sense of being lost in an inescapable labyrinth.14 Being familiar with the clinical characteristics and laboratory, electromyography, and neuroimaging markers of the main forms (Fig. 4) is helpful in diagnosis, and in many cases makes massively parallel sequencing of exomes and (costly) gene panels unnecessary. Identification of the specific mutation concerned is essential to establishing a prognosis and providing satisfactory treatment and proper genetic counselling. DNA repair defects, mitochondrial dysfunction, synaptic transmission disorders, loss of chaperone function, and metabolic pathway alterations are known to be involved in the pathogenesis of autosomal recessive ataxias67; however, no effective treatment is available for many of these conditions, with early diagnosis being the greatest priority.

The 4 pillars for diagnosing autosomal recessive cerebellar syndromes of probable degenerative nature. ABLP: abetalipoproteinaemia; AOA1: ataxia with oculomotor apraxia type 1; ARCA: autosomal recessive cerebellar ataxia; ARSACS: autosomal recessive spastic ataxia of Charlevoix-Saguenay; AT: ataxia-telangiectasia; AVED: ataxia with vitamin E deficiency; CX: cerebrotendinous xanthomatosis; FA: Friedreich ataxia; MIRAS: mitochondrial recessive ataxia syndrome; MSS: Marinesco-Sjögren syndrome; NPC: Niemann-Pick disease type C; RD: Refsum disease; SANDO: sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; TSD: Tay-Sachs disease; WD: Wilson disease.
None declared.
The author is grateful to Dr Ángel Ortega (Granada) and Dr Juan Manuel Pías-Peleteiro (Santiago de Compostela) for the image permissions granted.
Please cite this article as: Arias M. Claves para afrontar el reto diagnóstico de las heredoataxias recesivas. Neurología. 2019;34:248–258.
