




The term IgG4-related disease (IgG4-RD) encompasses a heterogeneous group of systemic diseases characterised by tumefactive lesions in the affected organs, with lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells distributed in a pattern of storiform fibrosis.1 Considering the different names given to this entity throughout history, its prevalence and incidence remain unknown. It generally manifests in the sixth or seventh decade of life and predominantly affects men (60%-80%).2 The pathophysiological mechanism remains unknown, although the most relevant theories propose an autoimmune or allergic phenomenon, with T-helper 2 cells reacting disproportionately to an as yet unidentified antigen.3 Clinical manifestations and forms of presentation are varied and may involve multiple organs; differential diagnosis includes various neoplastic, infectious, and inflammatory processes (Table 1).4 The central nervous system (CNS) is rarely affected, although certain neurological syndromes are closely associated with IgG4-RD, such as hypertrophic pachymeningitis (HP) and hypophysitis.5 We present 2 cases of probable IgG4-RD with neurological symptoms as the initial manifestation. The objective of our study is to analyse the different forms of neurological presentation of IgG4-RD, with the aim of increasing neurologists’ clinical suspicion in order to promptly establish effective treatment.
Clinical diagnostic criteria and syndromes included in the spectrum of IgG4-related diseases.
| Diagnostic criteria | Syndromes |
|---|---|
| 1. Clinical examination showing masses or diffuse enlargement of one or several organs2. High serum IgG4 levels (≥ 135mg/dL)3. Anatomical pathology study revealing:a. Pronounced lymphocytic and plasmacytic infiltrate, and fibrosisb. IgG4+ plasmacytic infiltrates: IgG4+/IgG+ cell ratio > 40%, and > 10 IgG4+ cells/high-power field | - Autoimmune pancreatitis- Mikulicz syndrome- Küttner tumour- Mediastinal fibrosis- Riedel thyroiditis- Retroperitoneal fibrosis- Mesenteric sclerosis- Multifocal fibrosis- Inflammatory pseudotumour- Eosinophilic angiocentric fibrosis- Multifocal fibrosclerosis- Periaortitis and polyarteritis nodosa- Inflammatory aortic aneurysm- Idiopathic hypocomplementemic tubulointerstitial nephritis with extensive tubulointerstitial deposits- Hypertrophic pachymeningitis- Hypophysitis |
Diagnosis: definitive: 1+2+3; probable: 1+3; possible: 1+2.
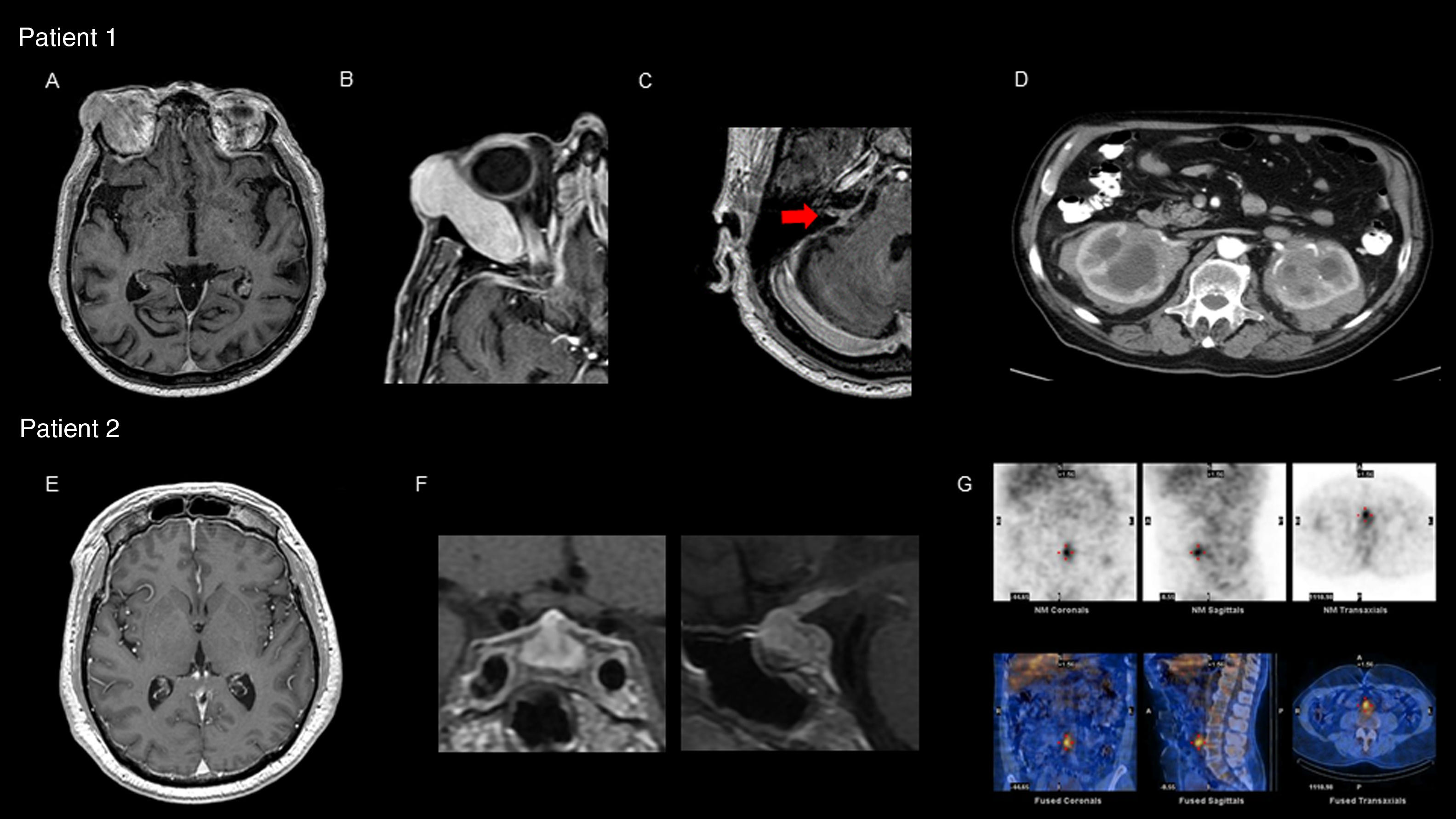
Our first patient was a 77-year-old man who was attended due to 6-year history of diplopia associated with chills and nocturnal sweating. The physical examination revealed right supraorbital tumour with proptosis and limited right ocular motility, bilateral anacusis, and right peripheral facial nerve palsy. A brain magnetic resonance imaging (MRI) scan showed pachymeningeal enhancement, predominantly located in the infratentorial region, and a tumour affecting the right lacrimal gland (Fig. 1A-C). Laboratory tests revealed an erythrocyte sedimentation rate of 60mm/h and C-reactive protein level of 34.21mg/L. Serum total IgG and IgG subtype 4 levels were normal. Cerebrospinal fluid analysis revealed high protein levels, with no microbial pathogens or neoplastic cells detected. An abdominal computed tomography (CT) scan showed enlarged kidneys due to infiltrative lesions (Fig. 1D). An anatomical pathology study of a renal biopsy specimen showed extensive lymphoplasmacytic infiltrate and positive immunostaining for CD138 and IgG4. We interpreted the case as probable IgG4-RD and started oral corticosteroid treatment and azathioprine, with no new clinical manifestations presenting. Laboratory results for inflammatory parameters normalised and the pachymeningeal enhancement resolved at 6 months of follow-up. However, bilateral anacusis remained as a sequela.
 brain MRI (gadolinium-enhanced T1-weighted sequence) showing diffuse pachymeningeal enhancement; (B) expansive right orbital lesion displacing the eye, showing homogeneous enhancement after contrast administration; (C) involvement of the acoustic-facial bundle (arrow); (D) abdominal CT scan with contrast: expansive infiltrative lesion affecting both kidneys, showing heterogeneous enhancement after contrast administration. Patient 2: (E) brain MRI (gadolinium-enhanced T1-weighted sequence) showing diffuse pachymeningeal enhancement; (F) diffuse enlargement and heterogeneous signal in the pituitary gland with pronounced homogeneous enhancement after contrast administration; (G) gallium scintigraphy: increased periaortic and interaortocaval uptake in images of the prevertebral retroperitoneum extending to the anterior part of the L4-L5 disc space.")
Patient 1: (A) brain MRI (gadolinium-enhanced T1-weighted sequence) showing diffuse pachymeningeal enhancement; (B) expansive right orbital lesion displacing the eye, showing homogeneous enhancement after contrast administration; (C) involvement of the acoustic-facial bundle (arrow); (D) abdominal CT scan with contrast: expansive infiltrative lesion affecting both kidneys, showing heterogeneous enhancement after contrast administration. Patient 2: (E) brain MRI (gadolinium-enhanced T1-weighted sequence) showing diffuse pachymeningeal enhancement; (F) diffuse enlargement and heterogeneous signal in the pituitary gland with pronounced homogeneous enhancement after contrast administration; (G) gallium scintigraphy: increased periaortic and interaortocaval uptake in images of the prevertebral retroperitoneum extending to the anterior part of the L4-L5 disc space.
Our second patient was a 58-year-old man who was attended due to 4-month history of headache, blurred vision, and dyschromatopsia. The physical examination revealed visual acuity of 20/70 in both eyes. A brain MRI scan revealed diffuse pituitary enlargement with contrast enhancement, associated with pachymeningeal enhancement in both frontal lobes and near the brainstem (Fig. 1E and F). Laboratory tests revealed an erythrocyte sedimentation rate of 70mm/h and C-reactive protein level of 44.4mg/L associated with panhypopituitarism, with normal serum IgG and IgG subtype 4 levels. Cerebrospinal fluid analysis revealed high protein levels, with no infectious or neoplastic signs. Findings from an abdominal CT scan and gallium scintigraphy were compatible with retroperitoneal fibrosis (Fig. 1G). An anatomical pathology study of a pituitary biopsy specimen showed extensive lymphoplasmacytic infiltrate and positive immunostaining for CD138 and IgG4. The patient was diagnosed with IgG4-RD; considering the severe involvement of both optic nerves, we started treatment with intravenous corticosteroids and cyclophosphamide combined with plasmapheresis, with an initially stable response. The patient continued treatment with azathioprine and after 20 months displayed no new clinical manifestations or visual sequelae. Inflammatory parameters normalised and the pituitary gland decreased in size. Panhypopituitarism was treated with hormone replacement therapy.
The most typical forms of CNS involvement in IgG4-RD are HP and hypophysitis, although orbital pseudomotor and pterygopalatine fossa fibrosis may also cause indirect nerve compression.4 HP frequently manifests with headache and cranial neuropathy, with compression of several structures of the nervous system or its vessels. MRI findings may reveal pachymeningeal involvement with a diffuse thickening or bulging mass.6 Hypophysitis manifests as headache and visual impairment and is associated with symptoms of hormone imbalance. The MRI scan may reveal diffuse thickening or the formation of masses in the pituitary gland and stalk. Both entities may manifest simultaneously.7 CNS involvement in IgG4-RD is included in the differential diagnosis of several processes: infection (neurosyphilis, tuberculosis, systemic mycoses), inflammation (granulomatosis with polyangiitis, giant-cell arteritis, rheumatoid arthritis, and neurosarcoidosis), cancer (meningeal carcinomatosis, lymphoma, meningioma, craniopharyngioma, adenoma, or pituitary germinoma), and even local benign tumours (ruptured Rathke cleft cyst).4,6,7
Multiple diagnostic criteria are used in daily clinical practice; however, they are not currently standardised. The most widely used combine clinical, laboratory, and anatomical pathology findings (Table 1)8,9: the presence of all 3 parameters is necessary to establish a definitive diagnosis of IgG4-RD. However, up to 50% of patients with a diagnosis confirmed by anatomical pathology study may present normal serum IgG4 levels10; therefore, presence of normal values should not determine the therapeutic approach, as in our 2 cases. Patients with greater disease activity present high circulating plasmablast counts in flow cytometry studies, even if serum IgG4 levels are normal. However, no study has validated their diagnostic usefulness,11 and anatomical pathology study of the affected organ remains the gold standard.12 No standardised treatment protocols are currently available. However, consensus exists on starting immunosuppressive treatment in patients with active symptomatic disease and those with subclinical manifestations that may potentially lead to severe and irreversible sequelae.13 Glucocorticoids are the first line drug used as the induction therapy and may be administered orally or even intravenously in patients with immediate functional risk, as was the case of our patient with severe visual impairment. Other immunosuppressive agents (azathioprine, mycophenolate, mercaptopurine, methotrexate, tacrolimus, cyclophosphamide) have been used as corticosteroid sparing agents, and even in monotherapy when corticosteroids are contraindicated, although their efficacy has not been demonstrated in prospective studies.13 As in other IgG4-mediated autoimmune disorders,14 rituximab has shown to be effective in IgG4-RD in retrospective studies, even in patients in whom treatment with corticosteroid-sparing agents is unsuccessful.13 After satisfactory induction therapy, some patients benefit from maintained immunosuppression (low doses of corticosteroids, corticosteroid-sparing agents, or rituximab), although the optimal duration has not yet been established and will depend on each particular case.13
In conclusion, although IgG4-RD is a systemic inflammatory disease affecting several organs, it may initially manifest with neurological symptoms only. It must be suspected in patients with PH and hypophysitis when other more frequent causes have been ruled out. Normal serum IgG4 levels should not rule out the diagnosis or determine the therapeutic approach. Anatomical pathology study of the affected organ remains the cornerstone of diagnosis. Early diagnosis and treatment are essential because once tissue fibrosis is established, sequelae are usually irreversible. Corticosteroids are the treatment of choice, and rituximab should be considered in severe or refractory cases.
Please cite this article as: Pastor Rueda JM, Alessandro L, Calandri IL, Cammarota Á. Manifestaciones neurológicas como presentación inicial de la enfermedad relacionada con IgG4. Neurología. 2020;35:276–279.
