



Creutzfeldt-Jakob disease (CJD) is a fatal, rapidly progressive neurodegenerative disease. Only 10% of cases are familial. These patients present a mutation in the gene encoding the prion protein, located on chromosome 20, resulting in either a methionine (M) or a valine (V).1,2 Few cases of familial CJD have been reported in Spain. We present 2 cases of familial CJD and provide the genealogy of a family with mutation E200K 129M/M.
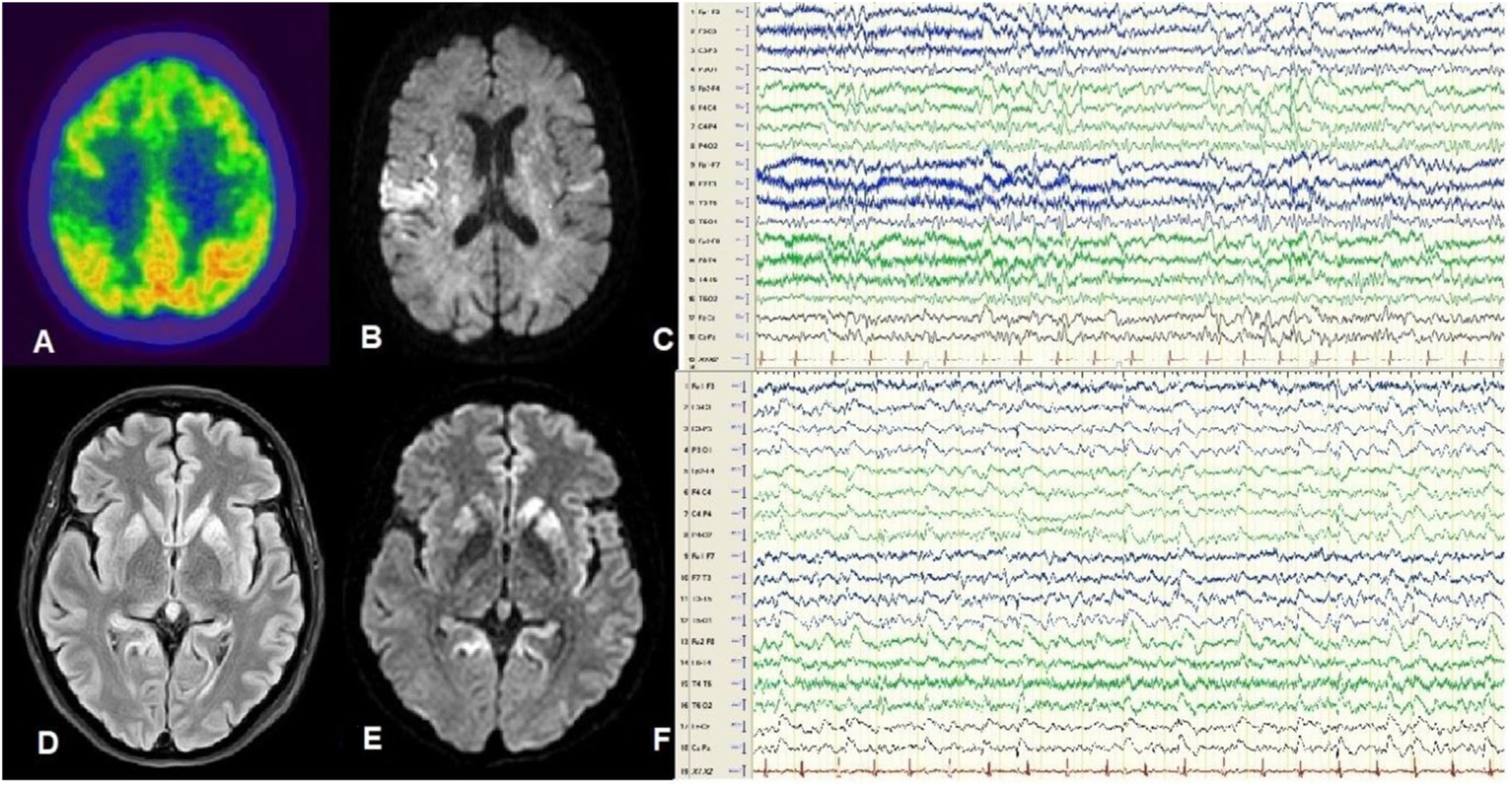
Patient 1Our first patient was a 59-year-old woman with no relevant history, who consulted due to cognitive impairment and gait disorder of one month’s progression. The patient developed akinetic mutism and died 2 months after symptom onset. Her clinical characteristics are summarised in Table 1. An 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) study performed 45 days after symptom onset revealed hypometabolism in the right parietal cortex and striatum (Fig. 1A), and an MRI study revealed bilateral hyperintensities predominantly in the insula and right parietal region on FLAIR and DWI sequences (Fig. 1B). An EEG study performed at 60 days recorded focal spike-and-wave discharges predominantly in the right parasagittal region (Fig. 1C). The patient tested positive for 14-3-3 protein in the cerebrospinal fluid, and a genetic study detected mutation E200K 129M/M. A post mortem study revealed spongiform changes in cortical and subcortical areas, predominantly in the left hemisphere.
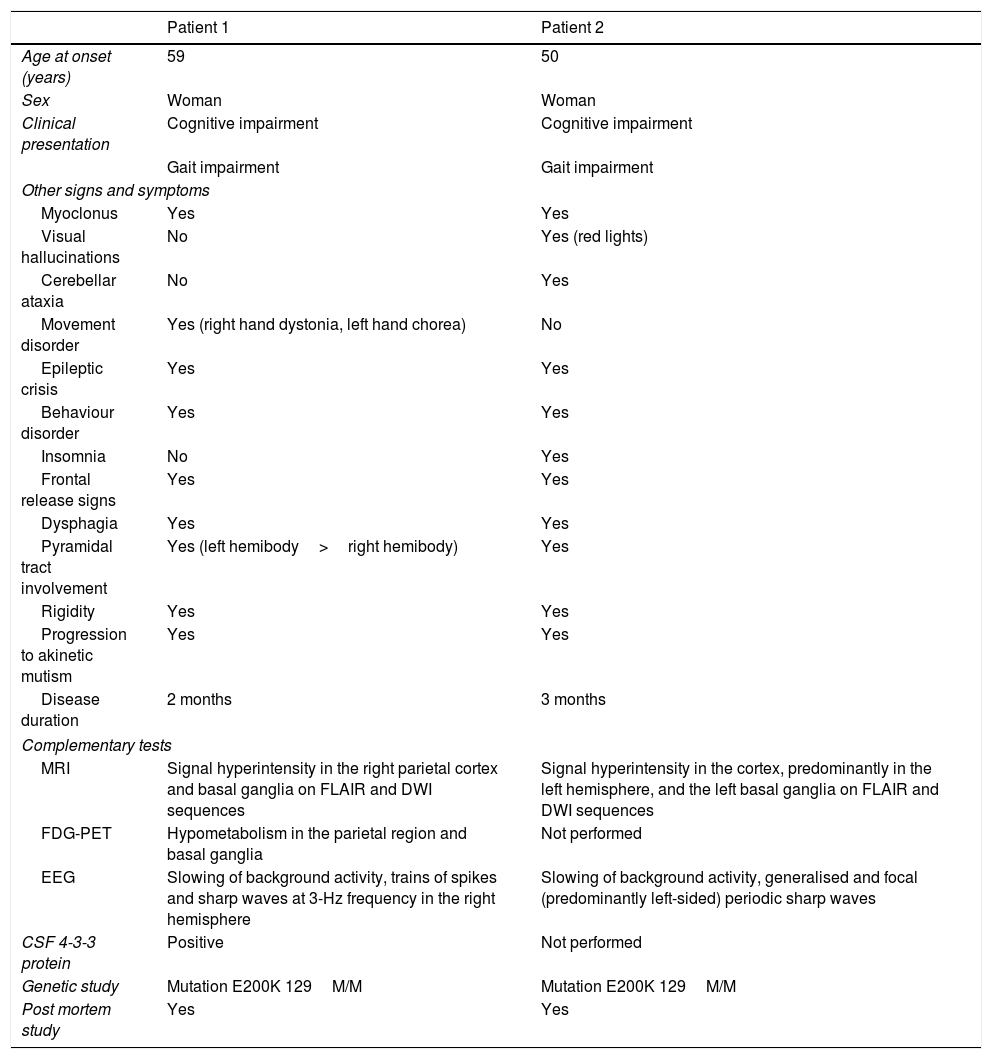
Clinical characteristics, semiological findings, and complementary test results from our 2 patients.
| Patient 1 | Patient 2 | |
|---|---|---|
| Age at onset (years) | 59 | 50 |
| Sex | Woman | Woman |
| Clinical presentation | Cognitive impairment | Cognitive impairment |
| Gait impairment | Gait impairment | |
| Other signs and symptoms | ||
| Myoclonus | Yes | Yes |
| Visual hallucinations | No | Yes (red lights) |
| Cerebellar ataxia | No | Yes |
| Movement disorder | Yes (right hand dystonia, left hand chorea) | No |
| Epileptic crisis | Yes | Yes |
| Behaviour disorder | Yes | Yes |
| Insomnia | No | Yes |
| Frontal release signs | Yes | Yes |
| Dysphagia | Yes | Yes |
| Pyramidal tract involvement | Yes (left hemibody>right hemibody) | Yes |
| Rigidity | Yes | Yes |
| Progression to akinetic mutism | Yes | Yes |
| Disease duration | 2 months | 3 months |
| Complementary tests | ||
| MRI | Signal hyperintensity in the right parietal cortex and basal ganglia on FLAIR and DWI sequences | Signal hyperintensity in the cortex, predominantly in the left hemisphere, and the left basal ganglia on FLAIR and DWI sequences |
| FDG-PET | Hypometabolism in the parietal region and basal ganglia | Not performed |
| EEG | Slowing of background activity, trains of spikes and sharp waves at 3-Hz frequency in the right hemisphere | Slowing of background activity, generalised and focal (predominantly left-sided) periodic sharp waves |
| CSF 4-3-3 protein | Positive | Not performed |
| Genetic study | Mutation E200K 129M/M | Mutation E200K 129M/M |
| Post mortem study | Yes | Yes |
CSF: cerebrospinal fluid; DWI: diffusion-weighted imaging; EEG: electroencephalography; FDG-PET: 18F-fluorodeoxyglucose positron emission tomography; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.
 Patient 1. FDG-PET revealed hypometabolism in the posterior insula and right parietal region (A). DWI MRI showed hyperintensities predominantly in the right insula (B). Awake EEG (10-20 system) revealed bilateral slow background activity and focal epileptiform activity in the right parasagittal region (C). D-F) Patient 2. FLAIR (D) and DWI MRI (E) revealed hyperintensities in the cortex and basal ganglia, predominantly in the left hemisphere. Awake EEG (10-20 system) showed generalised theta-delta activity as well as periodic sharp waves and slow spike-and-wave complexes, predominantly in the left hemisphere (F).")
A-C) Patient 1. FDG-PET revealed hypometabolism in the posterior insula and right parietal region (A). DWI MRI showed hyperintensities predominantly in the right insula (B). Awake EEG (10-20 system) revealed bilateral slow background activity and focal epileptiform activity in the right parasagittal region (C). D-F) Patient 2. FLAIR (D) and DWI MRI (E) revealed hyperintensities in the cortex and basal ganglia, predominantly in the left hemisphere. Awake EEG (10-20 system) showed generalised theta-delta activity as well as periodic sharp waves and slow spike-and-wave complexes, predominantly in the left hemisphere (F).
The patient’s father had died at age 63 years after a rapidly progressive neurodegenerative process lasting 3 months, but no autopsy was performed. Our patient’s relatives were informed about the diagnosis and advised to undergo a genetic study. Two of our patient’s 4 siblings (both women) and one of her 2 children (a man) presented mutation E200K 129M/M. One of the patient’s siblings (a man) refused genetic testing.
Patient 2Our second patient was a 50-year-old woman, a carrier of E200K 129M/M, and sister to our first patient. She presented progressive cognitive impairment and gait impairment of one month’s progression. She died in a state of akinetic mutism 3 months after symptom onset. Table 1 summarises the patient’s clinical characteristics. An MRI study performed at 60 days revealed bilateral cortical hyperintensities in the caudate and lenticular nuclei, predominantly in the left hemisphere, on FLAIR and DWI sequences (Fig. 1D and E). An EEG study revealed bilateral focal and generalised periodic sharp waves, predominantly in the left hemisphere (Fig. 1F). No additional complementary tests were performed, in accordance with the wishes of the patient and her relatives. A post mortem study revealed spongiform changes in cortical and subcortical regions, predominantly in the left hemisphere.
Few cases of familial CJD have been reported in Spain; family history of the condition has only been confirmed in 3 of the 12 reported cases of genetic origin.3,4 In 2007, Morgado-Linares et al.5 presented 3 cases of a family with mutation E200K 129M/M, the most frequent mutation in Europe.6
In both of our patients, MRI revealed bilateral, asymmetric hyperintensities in the cortex and basal ganglia on FLAIR and DWI sequences; these are characteristic findings in both sporadic and genetic CJD.1 Basal ganglia involvement is more frequent in patients with the E200K mutation, and has been correlated with shorter survival times.7,8
Neither of our patients showed periodic sharp wave complexes on EEG, a typical finding in CJD whose incidence is variable in patients with the E200K mutation (38%-75%).2,9 Both patients showed a correlation between the hemisphere with more marked cortical involvement on MRI and laterality of epileptiform activity, a well-known association in patients with the E200K mutation.9
As is observed in sporadic CJD, patient 1 presented a correlation between the regions displaying hypometabolism on FDG-PET and radiological and anatomical pathology findings.10 In fact, 51.5% of cases of sporadic CJD present a correlation between FDG-PET findings and cortical alterations on MR images, and 34%-80% present a correlation between FDG-PET and post mortem findings.11
Our 2 patients present similar clinical characteristics to those of other reported cases with the same mutation, and patients with the MM1 subtype, the most frequent subtype of sporadic CJD. Clinical differentiation between sporadic and familial CJD is not straightforward. Complementary tests help to characterise the disease and to distinguish between sporadic and familial forms. Genetic testing of the patient and their relatives is essential to prevent misdiagnosis of familial CJD as sporadic.
In conclusion, we present the cases of 2 sisters with familial CJD and mutation E200K 129M/M, from a family with at least 2 affected generations. We describe the topographic correlation between FDG-PET, MRI, EEG, and anatomical pathology findings in one patient, and between MRI, EEG, and anatomical pathology findings in the other.
Please cite this article as: Sánchez-Soblechero A, Ros AL, Gómez Roldós A, Montoya-Aguirre G, Massot-Tarrús A. Enfermedad de Creutzfeldt-Jakob familiar por mutación E200K. Correlación en RM, EEG, PET y neuropatología en una familia. Neurología. 2021;36:399–401.
