





la discapacidad intelectual se presenta hasta en el 18% de algunas poblaciones. Las causas son variantes genéticas puntuales y variantes en el número de copias detectadas por la secuenciación de segunda generación y el análisis cromosómico por micromatrices, respectivamente. Sin embargo, se tiene otras variantes, como las estructurales, repetición de trinucleótidos o trastornos de la impronta que solo son detectadas por otras pruebas específicas. La secuenciación de tercera generación tiene el potencial de detectar todas las variantes genéticas. El objetivo es determinar las ventajas de la secuenciación de tercera generación sobre la de segunda generación en pacientes con discapacidad intelectual.
Material y métodosbúsqueda sistemática mediante los tesauros MESH «discapacidad intelectual» y «secuenciación de segunda generación»; así como el término de «secuenciación de tercera generación».
Resultadosse seleccionaron 31 artículos; 9 utilizaron la secuenciación de tercera generación en pacientes con estudios genómicos previos y se encontró que el 40% tenían variantes estructurales. Los que emplearon la secuenciación de segunda generación (n = 22) determinaron mediante un metaanálisis, que detectaron variantes de un solo nucleótido y variantes en el número de copias en un 29,8 y 9,2% de los casos, respectivamente.
Conclusionesla secuenciación de tercera generación tiene la capacidad de encontrar variantes estructurales, disomías uniparentales, repetición de trinucleótidos y variantes de un solo nucleótido, lo cual permitiría tener una mayor probabilidad de establecer la etiología de la discapacidad intelectual. Sin embargo, se necesitan estudios con muestras más representativas no solo en quienes padecen de discapacidad intelectual, sino a nivel poblacional.
Some populations have an intellectual disability frequency of nearly 18%. Among, the diverse genetic causes are single nucleotide variants and copy number variations, detected with second-generation sequencing and chromosomal microarray analysis, respectively. Nevertheless, other variants such as structural variants, trinucleotide repeat or imprinting disorders, cannot be detected by these tests and require different specific techniques. Third-generation sequencing have a power of found all variants. The purpose is to stablish the benefits of using third generation sequencing above second-generation sequencing in the diagnosis of patient with intellectual disability.
Material and methodsA rapid systematic review was performed on the Medline using thesaurus terms MESH of “intellectual disability” and “second-generation sequencing”; as well as using the term “third-generation sequencing”.
Results31 articles were selected in total. Of those, nine used third-generation sequencing in patients with previously genomic test, and founded structural variants in 40% of cases, all these variants were corroborated with other gold standard tests. Twenty-two studies used second-generation sequencing (n = 22) and showed through metanalysis, that 29,8% and 9,2% of these cases are due a single nucleotide variant and copy number variations, respectively.
ConclusionsThird-generation sequencing can find structural variants, uniparental disomies, trinucleotide repeat and single nucleotide variation. Therefore, it would allow a broader and better study of the etiology of intellectual disability. Nevertheless, more research with larger representative samples in patients and healthy population is needed.
La discapacidad intelectual (DI), conocida previamente como retraso mental, es el trastorno del neurodesarrollo más frecuente1,2. El diagnóstico se debe realizar en los pacientes que puedan realizar la prueba psicológica correspondiente (usualmente mayores de 5 años) y aquellos menores de 18 años, y se define por presentar limitación en el funcionamiento intelectual, así como dificultades conceptuales, sociales y áreas prácticas de la vida1,3.
La prevalencia de la DI es variable, estimándose entre el 1–3, 3–12,9 y el 18,3% a nivel mundial, latinoamericano y países de bajos o medianos ingresos, respectivamente4–8. La DI tiene un impacto importante a nivel económico y se calcula que los costos de un paciente con DI (en EE. UU. y Europa) está entre 1 y 2 millones de dólares, siendo más costoso que la demencia y el cáncer8.
Las causas genéticas de la DI son diversas. Entre ellas se encuentran las variantes en un solo nucleótido (SNV del inglés single nucleotide variant), variantes en múltiples nucleótidos (MNV del inglés multiple nucleotide variant), variantes en el número de copias (CNV del inglés copy number variation), aneuploidías, repetición de trinucléotidos y los rearreglos complejos9.
La secuenciación nueva generación o NGS (next generation sequencing), o también conocida como de alto rendimiento, es capaz de determinar la secuencia de bases en una cadena de ADN o ARN de forma paralela (varias cadenas a la vez) y masiva de varias plantillas a partir de una sola muestra, obteniéndose una enorme cantidad de datos10.
Dentro de las tecnologías de NGS que se usan actualmente para dignóstico clínico se tiene a la secuenciación de segunda generación (SSG), el cual se caracteriza por hacer lecturas de segmentos cortos de ADN que luego son alineados a un genoma de referencia y empalmados para generar la secuencia completa. Hay varios tipos de SSG, como los de secuenciación de paneles (de decenas a cientos de genes), exoma o WES (whole exome sequencing) y genoma o WGS (whole genome sequencing)11,12. Pero a pesar de que esta tecnología es en la actualidad ampliamente utilizada en el campo clínico, presenta importantes desventajas, por ejemplo, el genoma al poseer regiones repetitivas, los fragmentos cortos generados por la SSG no se alinean eficientemente y provocan una mala lectura o espacios «no leídos»12. Además, las variantes de un solo nucleótido o pequeñas deleciones/inserciones pueden ser detectadas a través de esta prueba12. Mientras que las variantes estructurales largas u otras son más difíciles de detectar a través de la SSG12.
Considerando estos puntos en contra de la SSG, se ha desarrollado la secuenciación de tercera generación (STG), que secuencia nucleótido por nucleótido, sin amplificación y en tiempo real, obteniendo además fragmentos largos10,12. Asimismo, hace un menor uso de recursos bioinformáticos y obtiene menos sesgos que en la SSG provocados por la amplificación clonal que detecta señales de incorporación de bases durante el uso de la PCR10.
A la fecha, el número de enfermedades genéticas asociadas a la DI son más de 2.0009. Por lo que se ha planteado como primera prueba de elección en los pacientes con DI la secuenciación de WES13,14. En este sentido, esta podrá detectar SNV en el 31 y 53% de los pacientes con DI aislada y sindrómica respectivamente13. Por otro lado, la secuenciación de WGS tiene, además, la posibilidad de detectar no solo SNV, sino CNV, regiones de homocigosidad y rearreglos cromosómicos15. Es así que en los pacientes con DI la WGS puede encontrar variantes patogénicas o probablemente patogénicas entre 27 y 62% de los casos16–18.
Se ha reportado que la STG (también conocida como LRS o long-read sequencing), puede encontrar variantes en los pacientes con DI que tuvieron pruebas negativas previas, como WES, WGS, análisis cromosómicos por micromatrices o CMA (chromosomal microarray analysis) o metilomas19. Las variantes que detecta la STG incluye las SNV, CNV, rearreglos cromosómicos complejos y las repeticiones en tándem20.
El objetivo de esta revisión es la de mostrar las ventajas que podría tener la STG de encontrar variantes genéticas en los pacientes con discapacidad intelectual cuyos resultados son negativos cuando se realizan las pruebas genómicas de uso clínico actual, como WES, WGS o CMA.
Material y métodosRevisión rápida de la literatura sobre el uso de la SSG y STG en el diagnóstico de la discapacidad intelectual.
Criterios de elegibilidadCriterios de inclusión
- i.
Relacionados con el tema a tratar: empleo de secuenciación de segunda y tercera generación en los pacientes con discapacidad intelectual.
- ii.
Tipos de estudio:
- •
Revisiones sistemáticas, estudios observacionales, pruebas diagnósticas en los estudios con SSG.
- •
Revisiones sistemáticas, estudios observacionales, pruebas diagnósticas en los estudios, casos y serie de casos con STG.
- iii.
Año de publicación:
- •
Todos los estudios de secuenciación de tercera generación.
- •
SSG: 2017–2022.
- iv.
Idiomas: aquellos publicados en inglés y español.
Criterios de exclusión
Tipos de estudios: cartas al editor, editoriales, comentarios, fichas técnicas, reporte de casos (reportes de segunda generación), revisiones narrativas.
Pregunta PICO: ¿el uso de secuenciación de tercera generación detecta con mayor frecuencia variantes patogénicas que la prueba de segunda generación en pacientes con discapacidad intelectual?
Población: pacientes con discapacidad intelectual.
Exposición: STG.
Comparador: SSG.
Desenlace:
Mayor de detección de variantes patogénicas en un solo nucleótido a través de STG.
Mayor de detección de variantes patogénicas en múltiples nucleótidos a través de STG.
Detección de otras variantes patogénicas.
Fuentes de informaciónSe revisó la base de datos de MEDLINE/PUBMED. Adicionalmente, se exploró manualmente las publicaciones que citaron los artículos de interés, así como las publicaciones citadas de los mismos estudios.
Estrategia de búsquedaLa estrategia de búsqueda se basó en los términos MeSH (Medical Subject Headings), intellectual disability y high throughput DNA sequencing. El término de long-read sequencing, no se encontró como descriptor en MeSH o DeCS; sin embargo, se utilizaron todos los sinónimos que fueron encontrados manualmente en las diferentes publicaciones. Se organizó la búsqueda en 2 grupos finales (Material suplementario 1). Se analizó la literatura en inglés y español. La fecha de búsqueda fue el 17 de septiembre del 2022 y fue actualizada el 22 de septiembre de 2022. Los datos fueron trasladados a Rayyan® para su posterior tamizaje según lectura de título y resumen.
Riesgo de sesgoPara determinar el riesgo de sesgo y la calidad de los datos de las publicaciones con diseños observaciones SSGse usó la escala de Newcastle-Ottawa modificada por los autores, teniendo un puntaje máximo de 10. Y para los reportes de casos STG se empleó la escala modificada de Murad et al., teniendo un puntaje máximo de 621.
Análisis de datosDe cada uno de los estudios que realizaron la SSG se extrajeron las frecuencias absolutas y relativas de los pacientes con las siguientes variantes genéticas: SNV y CNV patogénicas o probablemente patogénicas. De estos reportes, se hizo un metaanálisis utilizando el programa Jamovi® v. 2.3.18.0, calculando la heterogeneidad, mediante I2 y haciendo el diagrama de bosque para ambas variantes genéticas. En aquellos que usaron la STG se realizó la frecuencia absoluta y relativa del total de los estudios.
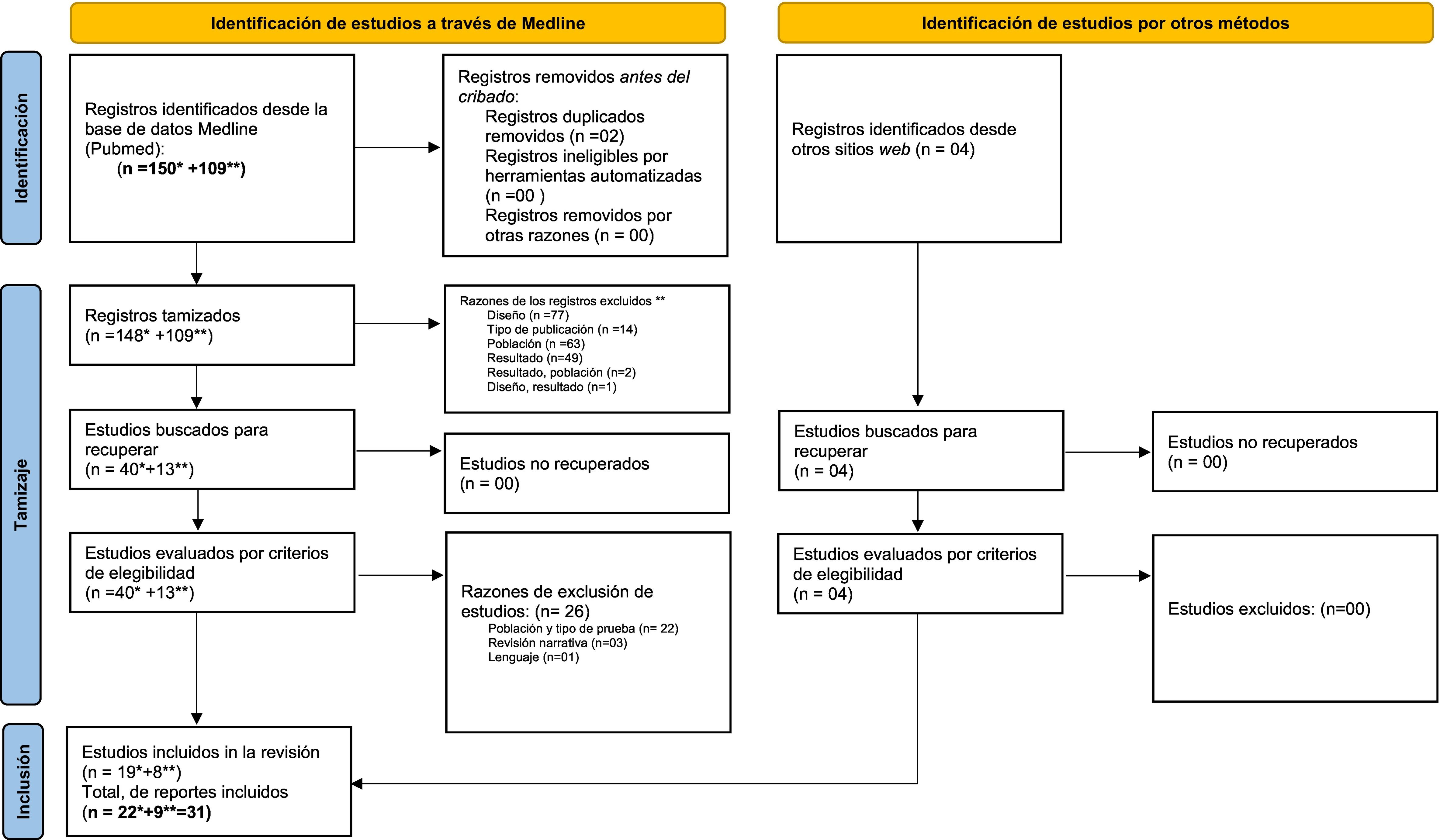
ResultadosSe encontraron 53 estudios según nuestra estrategia de búsqueda en Medline, al utilizar los criterios de elegibilidad y nuestra pregunta de investigación. Adicionalmente, se encontraron 4 artículos, a partir de las referencias que se incorporaron al análisis (fig. 1). Estas investigaciones fueron exportadas al programa de Zotero®.
![Selección de artículos científicos basado en PRISMA. *Secuenciación de segunda generación. ** Secuenciación de tercera generación. Extraído de: Page et al.[69].](https://static.elsevier.es/multimedia/11345934/0000003000000001/v1_202308071525/S1134593423000015/v1_202308071525/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcREUsqotiPBbMop0vPRR2H9/1VRsmKdKI5RBXQleOMR40fnNuecT5lKwQ3H/LN8vWCGwsQCTZ175RguMoY6i5qTfY+B2n7zyXB/DJp+V/6PRUrN3euVym6qgTsync3jqa+tSbUUGW5WYEosW27PP8djGCzWaSGsgmU71z8hQ2kfLlG9y7/vlOfssslJkSs+B3806ns0oGimSbhpba0PMzAYgDWyN0aOX+tjUq+vKRrpHWPY8SQmdBasuwQr7eatMk= "Selección de artículos científicos basado en PRISMA. *Secuenciación de segunda generación. ** Secuenciación de tercera generación. Extraído de: Page et al.[69].")
Selección de artículos científicos basado en PRISMA.
*Secuenciación de segunda generación.
** Secuenciación de tercera generación.
Extraído de: Page et al.[69].
Posteriormente, se realizó la lectura a texto completo y basados en los criterios de inclusión, quedaron 31 publicaciones, de las cuales 9 emplearon la STG y 22 la SSG. La exclusión de los 26 artículos restantes se procedió con base en el tipo de población estudiada y métodos de estudio (n = 22), revisión narrativa (n = 3) e idioma chino (n = 1) (material suplementario 2).
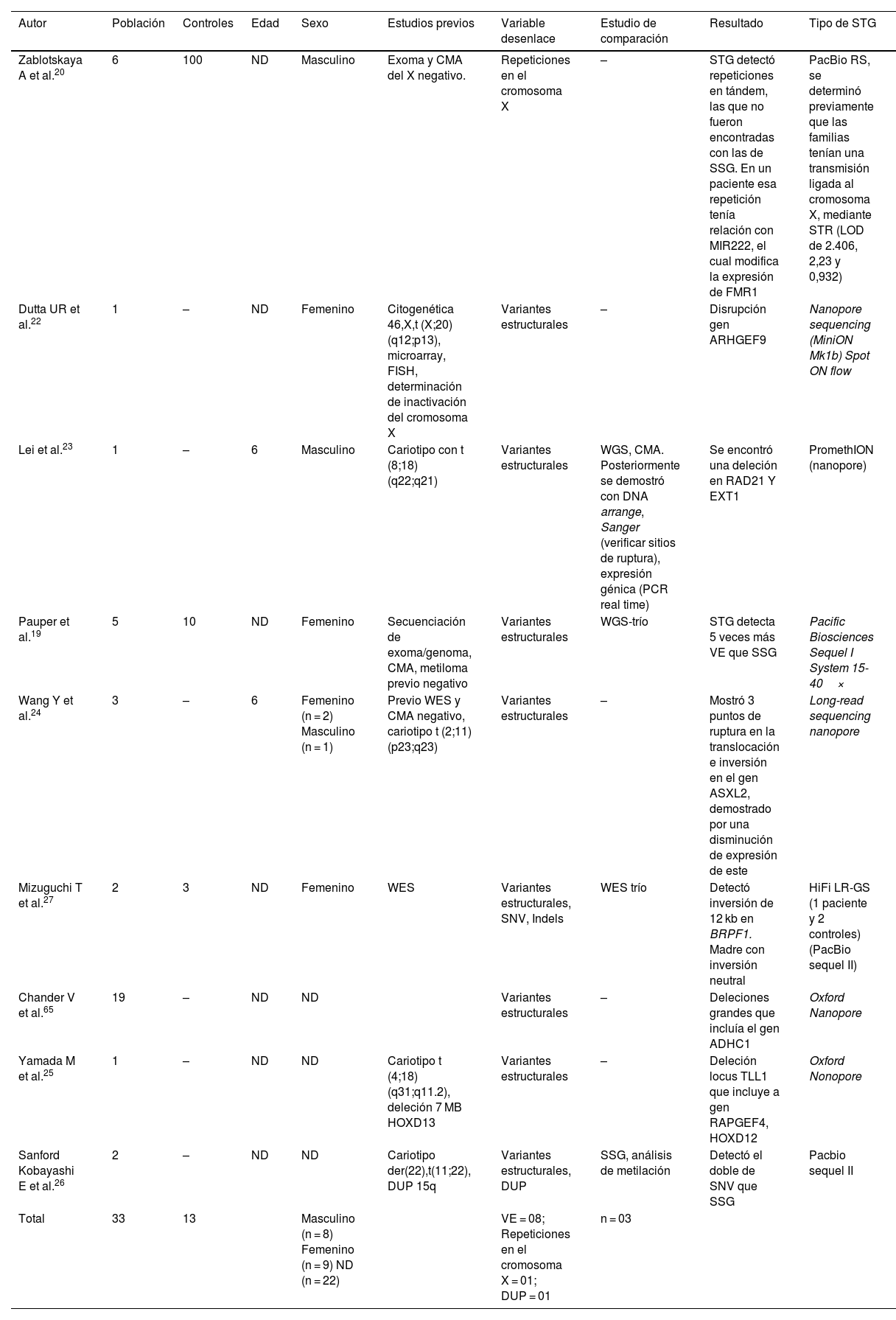
Secuenciación de tercera generaciónLos estudios que utilizaron la STG se realizaron en Japón (n = 2), Estados Unidos (n = 2), China (n = 2), Países Bajos (n = 1), Bélgica (n = 1) e India (n = 1). El número total de participantes fue de 153, de los cuales 40 fueron pacientes y 113 fueron controles sanos. En 3 estudios no se describió el sexo de los participantes (n = 22), y el sexo predominante de los participantes fue el femenino (52,9%). En 2 estudios se describió la edad de los pacientes (6 años). Los tipos de variantes genéticas que se analizaron fueron variantes estructurales (ej. translocaciones, CNV), SNV, disomías uniparentales y repetición de trinucleótidos. En 5 pacientes se pudo realizar la búsqueda simultánea de estas variantes y en la mayoría solo se buscó variantes estructurales (n = 29). La comprobación de la existencia de estas variantes fue realizada mediante diferentes técnicas como análisis cromosómico por micromatrices, secuenciación Sanger, expresión de ARN, Southern blotting, junction PCR, breakpoint mapping, RT-QPCR, análisis de metilación (tabla 1).
Características de los estudios en pacientes con discapacidad intelectual utilizado la secuenciación de tercera generación
| Autor | Población | Controles | Edad | Sexo | Estudios previos | Variable desenlace | Estudio de comparación | Resultado | Tipo de STG |
|---|---|---|---|---|---|---|---|---|---|
| Zablotskaya A et al.20 | 6 | 100 | ND | Masculino | Exoma y CMA del X negativo. | Repeticiones en el cromosoma X | – | STG detectó repeticiones en tándem, las que no fueron encontradas con las de SSG. En un paciente esa repetición tenía relación con MIR222, el cual modifica la expresión de FMR1 | PacBio RS, se determinó previamente que las familias tenían una transmisión ligada al cromosoma X, mediante STR (LOD de 2.406, 2,23 y 0,932) |
| Dutta UR et al.22 | 1 | – | ND | Femenino | Citogenética 46,X,t (X;20)(q12;p13), microarray, FISH, determinación de inactivación del cromosoma X | Variantes estructurales | – | Disrupción gen ARHGEF9 | Nanopore sequencing (MiniON Mk1b) Spot ON flow |
| Lei et al.23 | 1 | – | 6 | Masculino | Cariotipo con t (8;18) (q22;q21) | Variantes estructurales | WGS, CMA. Posteriormente se demostró con DNA arrange, Sanger (verificar sitios de ruptura), expresión génica (PCR real time) | Se encontró una deleción en RAD21 Y EXT1 | PromethION (nanopore) |
| Pauper et al.19 | 5 | 10 | ND | Femenino | Secuenciación de exoma/genoma, CMA, metiloma previo negativo | Variantes estructurales | WGS-trío | STG detecta 5 veces más VE que SSG | Pacific Biosciences Sequel I System 15-40× |
| Wang Y et al.24 | 3 | – | 6 | Femenino (n = 2) Masculino (n = 1) | Previo WES y CMA negativo, cariotipo t (2;11) (p23;q23) | Variantes estructurales | – | Mostró 3 puntos de ruptura en la translocación e inversión en el gen ASXL2, demostrado por una disminución de expresión de este | Long-read sequencing nanopore |
| Mizuguchi T et al.27 | 2 | 3 | ND | Femenino | WES | Variantes estructurales, SNV, Indels | WES trío | Detectó inversión de 12 kb en BRPF1. Madre con inversión neutral | HiFi LR-GS (1 paciente y 2 controles) (PacBio sequel II) |
| Chander V et al.65 | 19 | – | ND | ND | Variantes estructurales | – | Deleciones grandes que incluía el gen ADHC1 | Oxford Nanopore | |
| Yamada M et al.25 | 1 | – | ND | ND | Cariotipo t (4;18)(q31;q11.2), deleción 7 MB HOXD13 | Variantes estructurales | – | Deleción locus TLL1 que incluye a gen RAPGEF4, HOXD12 | Oxford Nonopore |
| Sanford Kobayashi E et al.26 | 2 | – | ND | ND | Cariotipo der(22),t(11;22), DUP 15q | Variantes estructurales, DUP | SSG, análisis de metilación | Detectó el doble de SNV que SSG | Pacbio sequel II |
| Total | 33 | 13 | Masculino (n = 8) Femenino (n = 9) ND (n = 22) | VE = 08; Repeticiones en el cromosoma X = 01; DUP = 01 | n = 03 |
CMA = Análisis cromosómico por micromatrices; DUP = Disomías uniparentales = 01; FISH = Hibridación in situ con fluorescencia; MB = megabases; STG = Secuenciación de tercera generación; SSG = Secuenciación de segunda generación; WES = Secuenciación de exoma completo; WGS = Secuenciación de genoma.
A 6 pacientes que tenían estudios citogenéticos previos que mostraban translocaciones balanceadas, se les realizó adicionalmente CMA o SSG, no encontrándose la etiología de la discapacidad intelectual22–25. Mediante el uso de la STG, se pudo encontrar variantes estructurales como deleciones o inversiones a nivel intragénico22–25. En un paciente, mediante la STG, se demostró una trisomía parcial del cromosoma 11 y 22 (síndrome Emanuel)26.
En el caso de 2 gemelas con DI y dismorfia, se hicieron WES-trío y al no encontrar variantes, se les realizó STG-trío en busca de SNV, variantes estructurales o inserciones/deleciones (indels), encontrando una VE ocasionada por una inversión en el gen BRPF1, el cual fue heredado de una inversión neutral materna27.
En 6 pacientes con DI, cuyo pedigrí sugería una herencia ligada al cromosoma X, se empleó CMA y exoma del cromosoma X, no encontrándose CNV ni repeticiones en tándem (RET). Luego de la STG se encontró una RET en el cromosoma X en uno de los pacientes que está relacionada con la expresión de MIR222, el cual a su vez está relacionado en la expresión del gen FMR1, por lo que probablemente esta variante estaría relacionado con el fenotipo descrito20.
En 15 pacientes (casos [n = 05] y controles familiares [n = 10]) con estudios previos de CMA, WES, metiloma sin resultados de variantes, se comparó la STG y la WGS en búsqueda de variantes estructurales. Ningún paciente presentó una variante relacionada con el fenotipo; no obstante, la STG encontró 5 veces más variantes estructurales que la SSG19.
Finalmente, en un paciente que tenía el diagnóstico de síndrome Prader-Willi con análisis de metilación positiva, la STG pudo determinar la presencia de una heterodisomía uniparental en la posición 15q11.226.
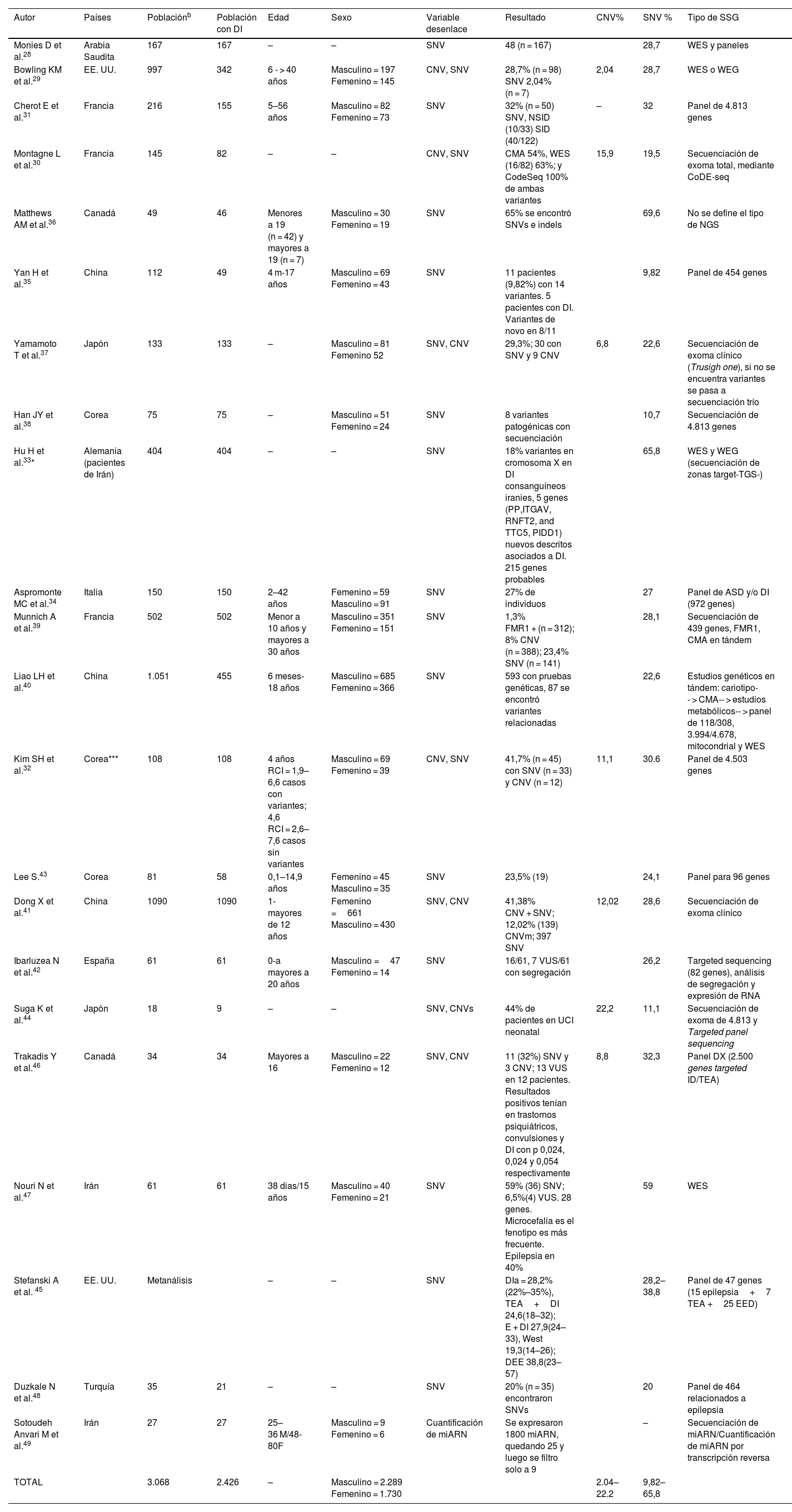
Secuenciación de segunda generaciónCon relación a los estudios que utilizaron la SSG (n = 22), uno era un metaanálisis y el resto eran observacionales. Las investigaciones se realizaron en mayor frecuencia en China (n = 3), Corea (n = 3), Canadá (n = 2), Japón (n = 2), Irán (n = 2), Francia (n = 2) y EE. UU. (n = 2). Las poblaciones estudiadas fueron heterogéneas (n = 5.516), con diagnósticos que no tenían necesariamente el fenotipo de DI. El número total de participantes con DI fue de 4.029, con edades fluctuantes entre pediátricas y adultos. Los tipos de pruebas que emplearon fueron desde la secuenciación de paneles que incluía un número de 47 genes hasta secuenciación de genoma-trío. En 6 estudios no se precisó el sexo de los participantes. Del total de los reportes que describieron el sexo, el 56,95% de los participantes fueron varones. La mayoría de los estudios determinaron SNV, excepto uno que utilizó la SSG como método para determinar la expresión de ARN. Adicionalmente, 7 estudios buscaron CNV (tabla 2)28–49.
Características de los estudios en pacientes con discapacidad intelectual utilizado la secuenciación de segunda generación (2017–2022)
| Autor | Países | Poblaciónb | Población con DI | Edad | Sexo | Variable desenlace | Resultado | CNV% | SNV % | Tipo de SSG |
|---|---|---|---|---|---|---|---|---|---|---|
| Monies D et al.28 | Arabia Saudita | 167 | 167 | – | – | SNV | 48 (n = 167) | 28,7 | WES y paneles | |
| Bowling KM et al.29 | EE. UU. | 997 | 342 | 6 - > 40 años | Masculino = 197 Femenino = 145 | CNV, SNV | 28,7% (n = 98) SNV 2,04% (n = 7) | 2,04 | 28,7 | WES o WEG |
| Cherot E et al.31 | Francia | 216 | 155 | 5–56 años | Masculino = 82 Femenino = 73 | SNV | 32% (n = 50) SNV, NSID (10/33) SID (40/122) | – | 32 | Panel de 4.813 genes |
| Montagne L et al.30 | Francia | 145 | 82 | – | – | CNV, SNV | CMA 54%, WES (16/82) 63%; y CodeSeq 100% de ambas variantes | 15,9 | 19,5 | Secuenciación de exoma total, mediante CoDE-seq |
| Matthews AM et al.36 | Canadá | 49 | 46 | Menores a 19 (n = 42) y mayores a 19 (n = 7) | Masculino = 30 Femenino = 19 | SNV | 65% se encontró SNVs e indels | 69,6 | No se define el tipo de NGS | |
| Yan H et al.35 | China | 112 | 49 | 4 m-17 años | Masculino = 69 Femenino = 43 | SNV | 11 pacientes (9,82%) con 14 variantes. 5 pacientes con DI. Variantes de novo en 8/11 | 9,82 | Panel de 454 genes | |
| Yamamoto T et al.37 | Japón | 133 | 133 | – | Masculino = 81 Femenino 52 | SNV, CNV | 29,3%; 30 con SNV y 9 CNV | 6,8 | 22,6 | Secuenciación de exoma clínico (Trusigh one), si no se encuentra variantes se pasa a secuenciación trío |
| Han JY et al.38 | Corea | 75 | 75 | – | Masculino = 51 Femenino = 24 | SNV | 8 variantes patogénicas con secuenciación | 10,7 | Secuenciación de 4.813 genes | |
| Hu H et al.33* | Alemania (pacientes de Irán) | 404 | 404 | – | – | SNV | 18% variantes en cromosoma X en DI consanguíneos iranies, 5 genes (PP,ITGAV, RNFT2, and TTC5, PIDD1) nuevos descritos asociados a DI. 215 genes probables | 65,8 | WES y WEG (secuenciación de zonas target-TGS-) | |
| Aspromonte MC et al.34 | Italia | 150 | 150 | 2–42 años | Femenino = 59 Masculino = 91 | SNV | 27% de individuos | 27 | Panel de ASD y/o DI (972 genes) | |
| Munnich A et al.39 | Francia | 502 | 502 | Menor a 10 años y mayores a 30 años | Masculino = 351 Femenino = 151 | SNV | 1,3% FMR1 + (n = 312); 8% CNV (n = 388); 23,4% SNV (n = 141) | 28,1 | Secuenciación de 439 genes, FMR1, CMA en tándem | |
| Liao LH et al.40 | China | 1.051 | 455 | 6 meses- 18 años | Masculino = 685 Femenino = 366 | SNV | 593 con pruebas genéticas, 87 se encontró variantes relacionadas | 22,6 | Estudios genéticos en tándem: cariotipo-- > CMA-- > estudios metabólicos-- > panel de 118/308, 3.994/4.678, mitocondrial y WES | |
| Kim SH et al.32 | Corea*** | 108 | 108 | 4 años RCI = 1,9–6,6 casos con variantes; 4,6 RCI = 2,6–7,6 casos sin variantes | Masculino = 69 Femenino = 39 | CNV, SNV | 41,7% (n = 45) con SNV (n = 33) y CNV (n = 12) | 11,1 | 30.6 | Panel de 4.503 genes |
| Lee S.43 | Corea | 81 | 58 | 0,1–14,9 años | Femenino = 45 Masculino = 35 | SNV | 23,5% (19) | 24,1 | Panel para 96 genes | |
| Dong X et al.41 | China | 1090 | 1090 | 1- mayores de 12 años | Femenino =661 Masculino = 430 | SNV, CNV | 41,38% CNV + SNV; 12,02% (139) CNVm; 397 SNV | 12,02 | 28,6 | Secuenciación de exoma clínico |
| Ibarluzea N et al.42 | España | 61 | 61 | 0-a mayores a 20 años | Masculino =47 Femenino = 14 | SNV | 16/61, 7 VUS/61 con segregación | 26,2 | Targeted sequencing (82 genes), análisis de segregación y expresión de RNA | |
| Suga K et al.44 | Japón | 18 | 9 | – | – | SNV, CNVs | 44% de pacientes en UCI neonatal | 22,2 | 11,1 | Secuenciación de exoma de 4.813 y Targeted panel sequencing |
| Trakadis Y et al.46 | Canadá | 34 | 34 | Mayores a 16 | Masculino = 22 Femenino = 12 | SNV, CNV | 11 (32%) SNV y 3 CNV; 13 VUS en 12 pacientes. Resultados positivos tenían en trastornos psiquiátricos, convulsiones y DI con p 0,024, 0,024 y 0,054 respectivamente | 8,8 | 32,3 | Panel DX (2.500 genes targeted ID/TEA) |
| Nouri N et al.47 | Irán | 61 | 61 | 38 dias/15 años | Masculino = 40 Femenino = 21 | SNV | 59% (36) SNV; 6,5%(4) VUS. 28 genes. Microcefalia es el fenotipo es más frecuente. Epilepsia en 40% | 59 | WES | |
| Stefanski A et al. 45 | EE. UU. | Metanálisis | – | – | SNV | DIa = 28,2% (22%–35%), TEA+DI 24,6(18–32); E + DI 27,9(24–33), West 19,3(14–26); DEE 38,8(23–57) | 28,2–38,8 | Panel de 47 genes (15 epilepsia+7 TEA +25 EED) | ||
| Duzkale N et al.48 | Turquía | 35 | 21 | – | – | SNV | 20% (n = 35) encontraron SNVs | 20 | Panel de 464 relacionados a epilepsia | |
| Sotoudeh Anvari M et al.49 | Irán | 27 | 27 | 25–36 M/48-80F | Masculino = 9 Femenino = 6 | Cuantificación de miARN | Se expresaron 1800 miARN, quedando 25 y luego se filtro solo a 9 | – | Secuenciación de miARN/Cuantificación de miARN por transcripción reversa | |
| TOTAL | 3.068 | 2.426 | – | Masculino = 2.289 Femenino = 1.730 | 2.04–22.2 | 9,82–65,8 |
CMA = Análisis cromosómico por micromatrices; CNV = Variantes en el número de copias; DI = Discapacidad intelectual; FISH = Hibridación in situ con fluorescencia; Indels = Inserciones/deleciones; NSID = Discapacidad intelectual no sindrómica; MB = megabases; SID = Discapacidad intelectual sindrómica; STG = Secuenciación de tercera generación; SSG = Secuenciación de segunda generación; SNV = Variantes de un solo nucleótido; WGS = Secuenciación de genoma; WES = Secuenciación de exoma completo. a Se estudiaron a 404 familias con más de dos afectados. b Población total. cNo se especifica el diagnóstico de los pacientes si presentaron retraso del desarrollo psicomotor o discapacidad intelectual.
La frecuencia de las variantes en un único nucleótido es muy variable y se encuentra entre el 10,2 y el65,8% de los pacientes con DI28–44,46–48. En un metaanálisis previo se encontró que la SSG detecta SNV en el 28,2% (IC 95%, 22–35%) de los pacientes con DI aislada; en el 24,6% (18–32%) con trastorno del espectro autista y DI; en el 27,9% (24–33%) con epilepsia y DI; en el 19,3% (14–26%) de pacientes con síndrome West y 38,8% (23–57%) de pacientes con encefalopatía epiléptica y del desarrollo45.
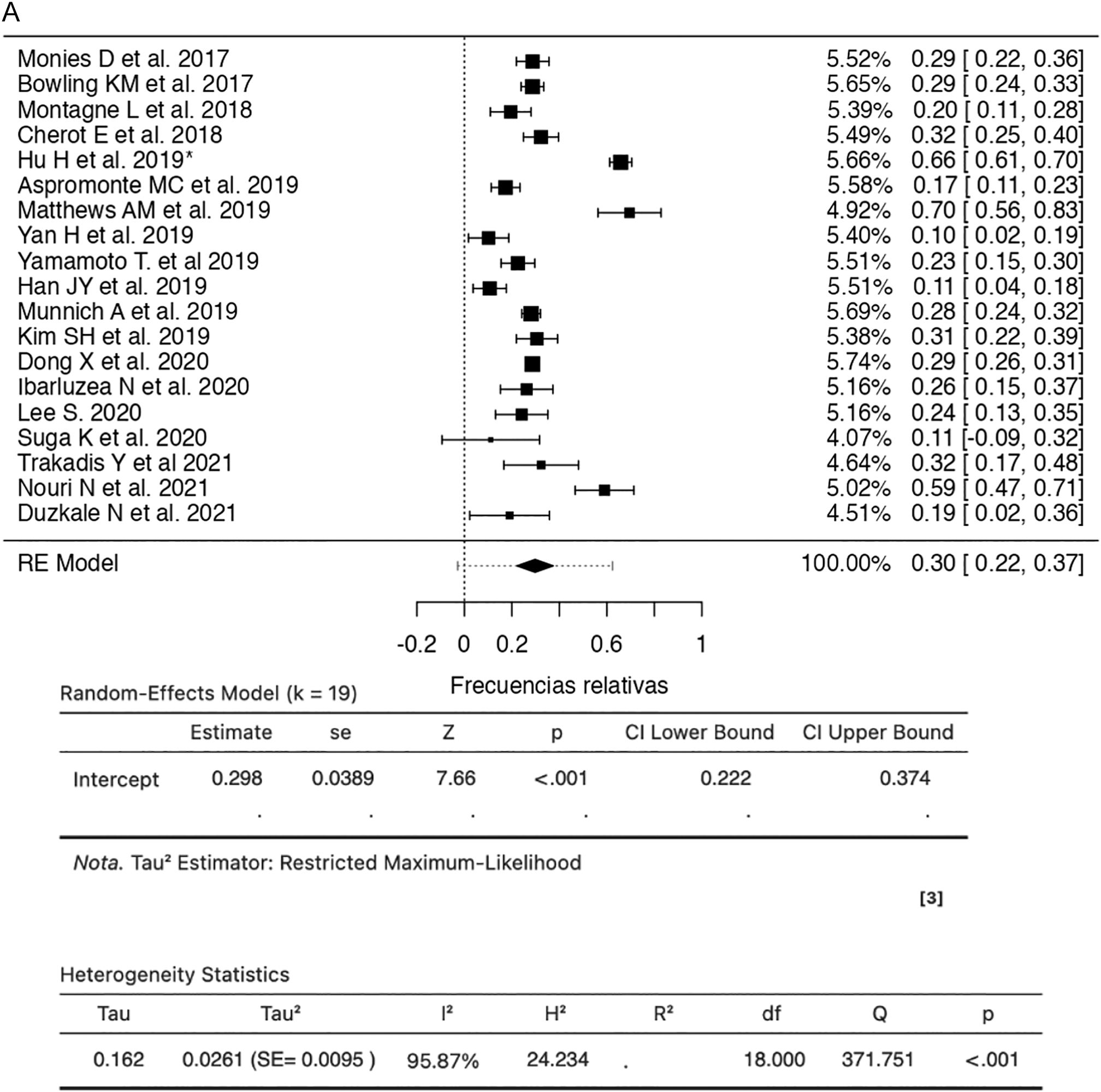
Al realizar el metaanálisis en el presente estudio, con relación a la detección de SNV mediante la SSG del presente estudio, encontramos que la frecuencia es de 29,8% (IC 95%; 22,2-37,4%, I2 = 95,8%) (fig. 2A).
. B. Reportes de secuenciación de segunda generación que determinan variantes en el número de copias en pacientes con discapacidad intelectual (2017–2022).")
. B. Reportes de secuenciación de segunda generación que determinan variantes en el número de copias en pacientes con discapacidad intelectual (2017–2022).")
Estudios de secuenciación de segunda generación que determinan las variantes en un solo nucleótido en pacientes con discapacidad intelectual (2017–2022).
B. Reportes de secuenciación de segunda generación que determinan variantes en el número de copias en pacientes con discapacidad intelectual (2017–2022).
Por otro lado, 7 investigaciones determinaron CNV mediante la SSG y encontraron variantes entre 2,02 y22,2% de los pacientes con DI29,30,32,37,41,44,46. Al proceder con el metaanálisis en el presente estudio, se encontró que la SSG puede detectar CNV en el 9,15% (IC 95%; 5,1-13,2%; I2 = 86,2%) (fig. 2B).
Adicionalmente, un estudio que utilizó SSG cuantificó la expresión de miARN en pacientes con el síndrome del X frágil, el cual detectó en 9 de ellos que puede servir no solo como diagnóstico, sino como biomarcador49.
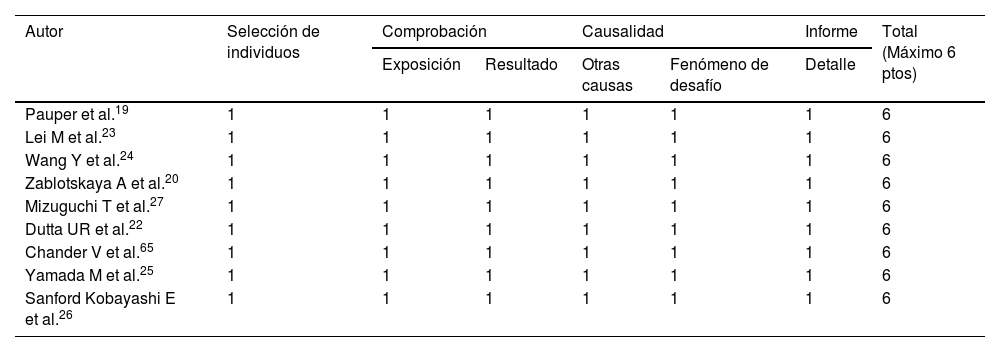
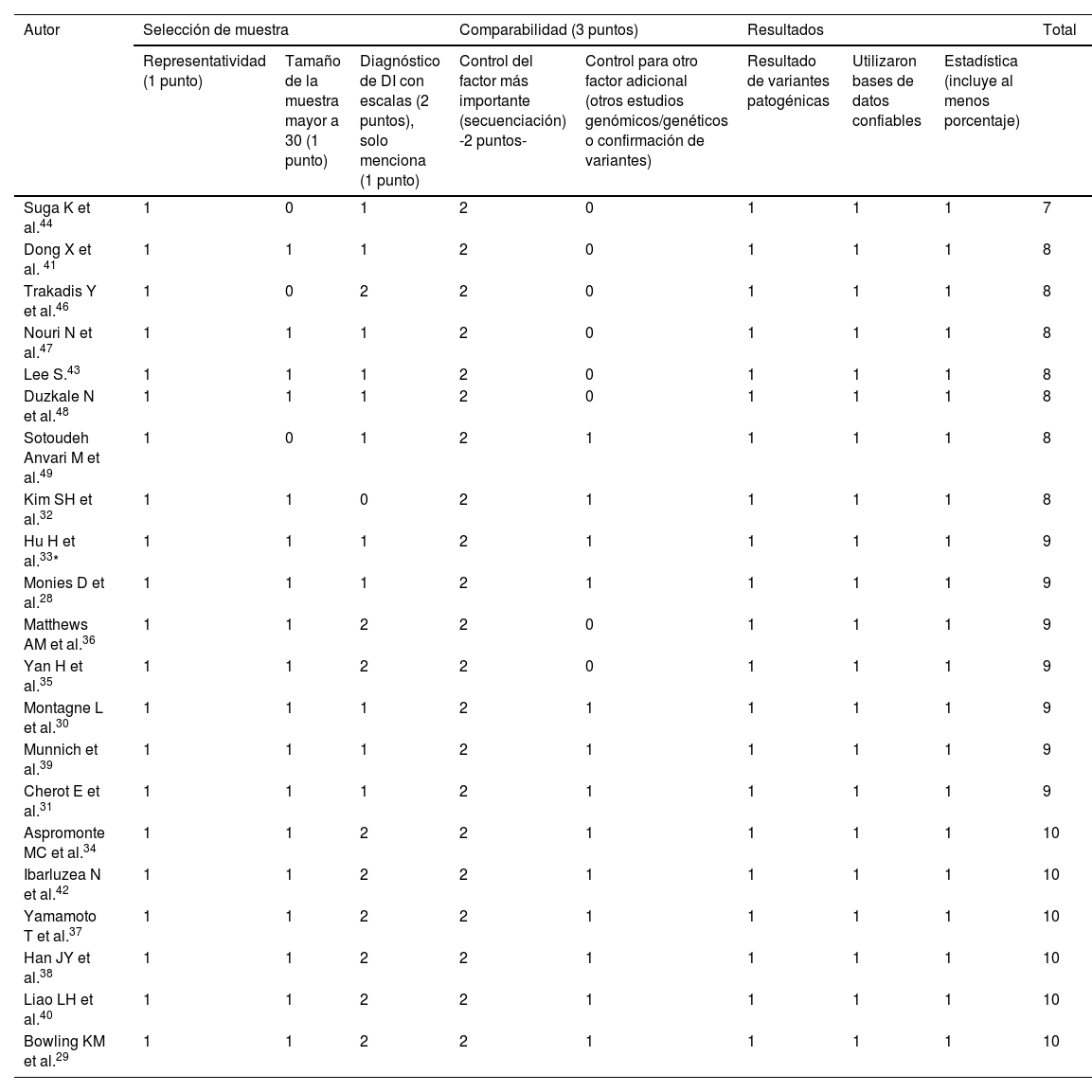
Riesgo de sesgo y calidad de la evidenciaLa calidad de los estudios que usaron STG se midió mediante la escala de Murad encontrando que todas las publicaciones tenían un riesgo de sesgo bajo (tabla 3). Con relación a los estudios de SSG la calidad se verificó mediante los criterios de Newcastle-Otawa que fueron modificados para el presente estudio, donde el puntaje máximo fue de 10, observando que uno obtuvo un puntaje de 7, y el resto obtuvo un puntaje mayor o igual a 8 (tabla 4).
Análisis de riesgo de sesgo de los estudios realizados en pacientes con discapacidad intelectual utilizando la secuenciación de tercera generación mediante la escala de Murad modificada
| Autor | Selección de individuos | Comprobación | Causalidad | Informe | Total (Máximo 6 ptos) | ||
|---|---|---|---|---|---|---|---|
| Exposición | Resultado | Otras causas | Fenómeno de desafío | Detalle | |||
| Pauper et al.19 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Lei M et al.23 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Wang Y et al.24 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Zablotskaya A et al.20 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Mizuguchi T et al.27 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Dutta UR et al.22 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Chander V et al.65 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Yamada M et al.25 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Sanford Kobayashi E et al.26 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
Calidad de los estudios realizados en pacientes con discapacidad intelectual utilizando la secuenciación de segunda generación (2017–2022), con criterios modificados de Newcastle-Otawa
| Autor | Selección de muestra | Comparabilidad (3 puntos) | Resultados | Total | |||||
|---|---|---|---|---|---|---|---|---|---|
| Representatividad (1 punto) | Tamaño de la muestra mayor a 30 (1 punto) | Diagnóstico de DI con escalas (2 puntos), solo menciona (1 punto) | Control del factor más importante (secuenciación) -2 puntos- | Control para otro factor adicional (otros estudios genómicos/genéticos o confirmación de variantes) | Resultado de variantes patogénicas | Utilizaron bases de datos confiables | Estadística (incluye al menos porcentaje) | ||
| Suga K et al.44 | 1 | 0 | 1 | 2 | 0 | 1 | 1 | 1 | 7 |
| Dong X et al. 41 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 8 |
| Trakadis Y et al.46 | 1 | 0 | 2 | 2 | 0 | 1 | 1 | 1 | 8 |
| Nouri N et al.47 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 8 |
| Lee S.43 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 8 |
| Duzkale N et al.48 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 8 |
| Sotoudeh Anvari M et al.49 | 1 | 0 | 1 | 2 | 1 | 1 | 1 | 1 | 8 |
| Kim SH et al.32 | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 1 | 8 |
| Hu H et al.33* | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 9 |
| Monies D et al.28 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 9 |
| Matthews AM et al.36 | 1 | 1 | 2 | 2 | 0 | 1 | 1 | 1 | 9 |
| Yan H et al.35 | 1 | 1 | 2 | 2 | 0 | 1 | 1 | 1 | 9 |
| Montagne L et al.30 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 9 |
| Munnich et al.39 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 9 |
| Cherot E et al.31 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 9 |
| Aspromonte MC et al.34 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
| Ibarluzea N et al.42 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
| Yamamoto T et al.37 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
| Han JY et al.38 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
| Liao LH et al.40 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
| Bowling KM et al.29 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 10 |
La SSG, que va desde los análisis de paneles hasta la secuenciación de genoma completo, se ha convertido en los últimos años en una herramienta más accesible, lo cual ha revolucionado el diagnóstico etiológico50.
Sin embargo, la frecuencia de detección de las variantes en las enfermedades genéticas, en general, ha permanecido casi constante51,52. Esto puede deberse a varios factores, como que la enfermedad evaluada no tenga un origen genético, que la variante genética hallada no pueda ser interpretada debido a la falta de información o que la causa molecular de la enfermedad no pueda ser detectada por la técnica utilizada26.
Es así que existe un grupo de pacientes con DI cuyos resultados de WES, WGS o CMA son negativos, a pesar de tener por ejemplo datos clínicos y un tipo de herencia determinado que se puede determinar por el número o el sexo de pacientes afectados, el número de generaciones comprometidas o el antecedente de consanguinidad, lo cual induce a plantear que la etiología de la discapacidad intelectual sería por la presencia de una variante genética. En ese sentido, en la actualidad existen herramientas que podrían ayudar en encontrar la etiología, mediante, por ejemplo, el uso de la secuenciación de ARN. Sin embargo, la disponibilidad del tejido afectado (ej. biopsia cerebral) la hace poco factible26.
La longitud de los reads se refiere al número de pares de bases (pb) secuenciados en un fragmento de ADN. Cuando se utiliza la SSG, estos fragmentos son cortos. Luego de la secuenciación, las regiones que se superponen son utilizadas para ensamblar los reads y alinearlos a un ADN de referencia, reconstruyendo así la cadena de ADN. Por ejemplo, la data obtenida con una plataforma Illumina® posee reads de 150 pb y puede alinearse con éxito aproximadamente el 84% del genoma humano53. Sin embargo, existen regiones repetidas que son fragmentos de 150 pb, los cuales corren el riesgo de mapearse a múltiples posiciones, y por lo tanto, son normalmente excluidas del análisis54. Estos territorios genómicos son llamados como «regiones muertas» para la SSG, incluyendo a las zonas repetidas (short tadem repeats o STR, regiones alfoides que componen los centrómeros), zonas de baja complejidad (ej. elementos ALU), o regiones duplicadas (ej. seudogenes). Además, la secuenciación de Illumina® es insensible a variaciones estructurales mayores a 50 pb, donde se estima que el 75% de los eventos, en su mayoría inserciones, son obviados55. Es evidente entonces que la SSG posee limitaciones intrínsecas debido a su propio desarrollo tecnológico.
Por otro lado, se tiene que la STG (también conocida como Long-Read Sequencing o LRS), que está basada en que los reads sean fragmentos largos de hasta más de 100 veces en comparación a la SSG (mayor a 10 Kpb hasta por encima de 1 Mb)56.
Es así que la STG está mostrando su potencialidad para detectar un mayor número de variantes con relación a la de segunda generación. En ese sentido, la STG tiene una alta fidelidad para las pequeñas variantes y puede identificar casi todas las variantes halladas por la SSG. Así mismo, tiene la habilidad de establecer la presencia de variantes estructurales, particularmente aquellas mediadas por elementos repetitivos o de baja complejidad (ej. elementos ALU). Además, detecta defectos en la metilación genómica, y por ende puede servir como una herramienta diagnóstica y funcional. Y por último es su capacidad de detectar eficientemente variantes en las «zonas muertas», lo cual se puede realizar con SSG solo si se aplican complejos métodos bioinformáticos, extensas bases de datos para eliminar falsos positivos y numerosos métodos ortogonales para confirmar las variantes26.
En la presente revisión rápida, se observó que en la cohorte de pacientes con DI que empleó la STG, todos tenían previamente o durante las evaluaciones análisis genómicos que no contribuían para el diagnóstico. La mayoría de los estudios en el que se utilizó la STG encontraron el cambio genético relacionado a la DI, siendo el más frecuente las variantes estructurales (40%)22–26. En este aspecto, se ha observado que el número de variantes estructurales encontradas en los pacientes es más frecuente cuando se usa la STG, que la SSG[19]. Y en aquellos con alteraciones cromosómicas estructurales aparentemente balanceadas, se detectó las variantes estructurales submicroscópicas en los sitios de ruptura y unión22–25. En este sentido, es muy probable que muchas de las variantes estructurales sean submicroscópicas (ej. inserciones, inversiones, translocaciones), las cuales no podrán ser detectadas mediante las pruebas de rutina disponible a la fecha57. En este grupo de enfermedades es donde justo tomaría protagonismo la STG, tal como se demostró en el paciente con una inversión patogénica en el gen BRPF127.
El síndrome del X frágil se observa en el 2,4% de las personas con DI y se considera la segunda causa relativa más frecuente después del síndrome de Down. Este síndrome es causado por una expansión de tripletes y esta condición se detecta mediante pruebas específicas y no son descubiertas por las pruebas genómicas como CMA o WGS58,59. La STG, adicionalmente, es capaz de detectar el número de repeticiones de los trinucleótidos CGG del gen FMR1 en portadores, incluido el de poder detectar el número de repeticiones AGG dentro del tándem de CGG, lo cual conferiría una ventaja adicional al estudio tradicional, que sería el de ofrecer no solo la posibilidad de diagnóstico, sino la estratificación del fenotipo60–63. En este sentido, tendría la capacidad de detectar segmentos en el genoma que presenten RET, las cuales podrían estar relacionadas a DI20.
Otra fortaleza del uso de la STG es el poder localizar variantes cromosómicas (ej. CNV o rreareglos complejos)26 o disomías uniparentales26 y lógicamente una mayor capacidad de encontrar SNV19.
Las limitaciones que tienen las investigaciones que utilizaron la STG es que ninguno calculó la exactitud diagnóstica, utilizando por ejemplo sensibilidad, especificidad o los valores predictivos; lo cual se debió principalmente al diseño y al número pequeño de participantes en cada estudio. Por otro lado, la STG no se empleó como primera prueba de elección, lo cual no permitió una comparación con la SSG ni mostrar las ventajas que son principalmente el de determinar otras variantes en «sitios muertos»64. Sin embargo, en todos los estudios de los pacientes con DI que se realizaron la STG comprobó la existencia de las variantes encontradas con otras técnicas gold standard, destacando así la buena calidad y el bajo riesgo de sesgo en todos los reportes19,20,22–27,65.
Con relación al metaanálisis del uso de SSG en pacientes con DI, las proporciones de SNV que se encontraron fueron similares a un estudio previo (29,2% vs. 28,6%)45. Una limitación de los estudios es que los criterios de selección no fueron similares y no se encontró una clasificación de la DI de forma uniforme según severidad o de diagnósticos asociados. Además, las pruebas de SSG utilizadas van desde el análisis de exones de algunas decenas hasta miles de genes, o la inclusión de las regiones intrónicas (secuenciación de genoma), lo cual muestra la gran heterogeneidad de los estudios.
Se observó también que la SSG mostró una capacidad de detectar CNV, por lo que según este trabajo, muestra que esta tecnología alcanzaría a establecer la etiología en el 9,2% de los pacientes con DI. No obstante, aún no conseguiría la frecuencia de detección que tiene el análisis cromosómico por micromatrices que está ≈ 30%66.
Se tiene que precisar que dependiendo del tipo de tecnología de secuenciación, donde los productos de estos segmentos secuenciados son cortos o largos, estos deberán ser comparados con genomas de referencia distintos. El genoma de referencia actual, GRCh38, está incompleto, ya que no contiene regiones heterocromáticas como los centrómeros y algunos segmentos idénticos duplicados (seudogenes). Con la tecnología SSG, esto no era un problema, pero con la STG se corre el riesgo de secuenciar fragmentos y no tener una referencia a la cual alinearlos67. Existe un nuevo genoma de referencia, T2T-CHM13, que contiene más de 240 Mbp de secuencia adicional, incluyendo todos los centrómeros, duplicaciones segmentales y los brazos cortos de los cromosomas acrocéntricos67. Este nuevo ensamble ya ha sido utilizado en estudios que confirman su utilidad en conjunto con tecnologías de STG68.
A pesar de que la STG tiene una mejor posibilidad de encontrar todas las variantes genéticas sobre la SSG y sus riesgos de sesgo y calidad de las diferentes publicaciones son adecuados, no son equiparables ya que los reportes que utilizaron la STG y SSG son estudios de casos y transversales, respectivamente.
Una limitación inherente al presente estudio es que al momento de realizar la búsqueda de manera sistemática, hemos encontrado que el descriptor relacionado a la STG no se encuentra en MESH ni DECS, y por otro lado la única base de datos fue la de Medline.
Considerando todo lo expuesto, estamos de acuerdo con los autores de un estudio previo, quienes concluyeron que la STG puede ser un examen de primera línea en laboratorios que no tienen una alta capacidad de aplicación de herramientas bioinformáticas o equipos de laboratorio26. Esto, basado adicionalmente, en que la calidad de todos los estudios es alta cuando se usó la STG. En el mismo sentido, no estaría contraindicado el uso de SSG en pacientes con DI, ya que observamos que la calidad de los estudios en su mayoría fue alta y debería seguir siendo la primera prueba de elección en este grupo de pacientes. Y el uso de CMA para la detección de CNV debiera aún ser la prueba complementaria, en aquellos pacientes con DI que tuvieron la secuenciación negativa.
Además, debido a las ventajas que posee la STG, las cuales fueron previamente descritas, se reduciría la cantidad potencial de exámenes adicionales necesarios para llegar al diagnóstico definitivo del paciente, si fuese un diagnóstico molecular, ahorrando tiempo y esfuerzo de parte del laboratorio, las plataformas y los médicos.
Son interesantes las ventajas que se describen para la tecnología STG frente a la SSG en el diagnóstico de la DI. Sin embargo, el hallazgo de un número mayor de variantes, si bien abre muchas puertas a la investigación, no tiene la misma repercusión en la clínica debido a la poca información que existe hasta la fecha acerca de la relación de causalidad entre cada variante y su potencial cuadro clínico. Es decir, se necesitan aún más estudios para saber el verdadero impacto que tendría la tecnología de STG en el diagnóstico de la DI y cuál es la relevancia clínica de estas nuevas variantes.
FinanciaciónNinguna.
AutoríaHugo Abarca participó en todas las fases de la investigación. Flor Vásquez participó en la introducción, interpretación de los resultados, discusión y la aprobación final del manuscrito. Este artículo es original.
AgradecimientosA las Doctoras Yamilee Hurtado y Marta Martina, por su enorme apoyo como guía en el diseño y en la redacción del manuscrito.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Se puede consultar material adicional a este artículo en su versión electrónica disponible en https://doi.org/10.1016/j.psiq.2023.100392.
Material suplementario