



Pheochromocytomas are adrenal paragangliomas. Potentially malignant, these tumors have a low incidence but clear importance. They can appear in various hereditary syndromes, especially in von Hippel-Lindau syndrome, multiple endocrine neoplasia-2 (MEN2), and familial paraganglioma syndromes. In sporadic cases, underlying genetic alterations are often found, and these findings are changing our understanding of the disease.
Although these tumors can manifest with a characteristic clinical presentation, in 13.1%–57.6% of cases, it is the radiologist who first suggests the diagnosis, indicating analyses for catecholamines or nuclear medicine examinations.
Radiologists should suspect a pheochromocytoma on detection of a well-delimited adrenal mass with rapid, intense enhancement that typically shows cystic and hemorrhagic phenomena, high T2 signal intensity, and the absence of macroscopic or microscopic lipids. The behavior in diffusion-weighted imaging usually does not provide very useful information. Approximately one-third of lesions show late washout similar to that seen with adenomas on CT. Percutaneous puncture should be avoided to avoid the risk of unleashing a severe hypertensive crisis.
El feocromocitoma es un paraganglioma adrenal potencialmente maligno, con baja incidencia, pero relevancia evidente. Puede aparecer en varios síndromes hereditarios, especialmente von Hippel-Lindau, neoplasia endocrina múltiple 2 y paraganglioma familiar. En casos esporádicos subyacen también frecuentemente alteraciones genéticas que están cambiando el paradigma de la enfermedad.
Aunque puede tener una presentación clínica característica, en un 13,1−57,6% de los casos es el radiólogo el primero en sugerirlo, indicando determinaciones analíticas de catecolaminas o exploraciones de Medicina Nuclear.
Debe sospecharse ante una masa adrenal bien delimitada con realce intenso y rápido, mostrando característicamente fenómenos quísticos y hemorrágicos, alta señal en T2 y ausencia de lípidos macro o microscópicos. El comportamiento en difusión no suele aportar información muy relevante. Aproximadamente un tercio presentan lavado tardío similar al del adenoma en TC. Debe evitarse su punción percutánea ante su sospecha, por el riesgo de desencadenar una crisis hipertensiva grave.
Since the 2022 World Health Organisation update, pheochromocytomas are considered potentially malignant adrenal paragangliomas.1,2 They arise from neural crest-derived precursor cells in the adrenal medulla which are capable of secreting various catecholamines, including epinephrine.
Extra-adrenal paragangliomas (EAPGL) can be sympathetic (commonly abdominal and also secretors, although not epinephrine) or parasympathetic (mostly cervical and non-secreting).1–3 Sympathetic EAPGL can settle in any region of the retroperitoneal sympathetic chain (especially in the organ of Zuckerkandl, a mass of chromaffin cells arranged around the abdominal aorta and particularly concentrated between the origin of the inferior mesenteric artery and the aortic bifurcation), in the mediastinum or in the bladder, where they can cause clinical episodes due to the release of catecholamines during urination. Radiologists play a fundamental role in diagnosis and surgical planning.
Epidemiology and clinical featuresPheochromocytomas are one of the potentially curable causes of hypertension, although they occur in less than 0.2% of hypertensive patients. The annual incidence is 0.8/10,000, although they are probably under-diagnosed (in a series of post-mortem examinations, 50% had not manifested clinically).4 There are no significant differences according to gender.5 Mean age is 47, and the mean size at diagnosis is 49 mm.
Although imprecise, the "rule of 10" memory aid can be useful: metastatic (2–10% depending on the series; EAPGL up to 20–25%); bilateral; familial (up to 40% in some studies); extra-adrenal; and without hypertension.
Approximately 50% are symptomatic, due to secretion of catecholamines, with paroxysmal symptoms of headache (90% of symptomatic patients), sweating (60–70%) and tachycardia. They can induce takotsubo-like cardiomyopathy. Episodic hypertension accompanies these symptoms in half of cases and sustained hypertension in 35–45%.3,5–7 Some 5–15% do not have hypertension, with that proportion increasing with the increase in the number of cases diagnosed incidentally through radiological imaging (13.1−57.6%).
n 2017 staging for EAPGL was included in the eighth edition of the American Joint Committee on Cancer Staging Manual and Handbook (Table 1). Predictors of metastatic risk are: size >5 cm; extra-adrenal location (double risk of death); and germline mutation in the succinate dehydrogenase B subunit (SDHB) gene (generally in EAPGL). Three histopathological metastasis risk scales stand out, with good negative predictive value1,2: Pheochromocytoma of the Adrenal Gland Scaled Score (PASS); Grading System for Adrenal Pheochromocytoma and Paraganglioma (GAPP); and Composite Pheochromocytoma/Paraganglioma Prognostic Score (COPPS). Some studies also point to infiltrative and invasive radiological behaviour as a predictor of malignancy.
Cancer staging of pheochromocytomas and paragangliomas according to the American Joint Committee on Cancer (AJCC).
| Primary tumour: T | Regional lymph nodes. N | Distant metastasis: M | |
|---|---|---|---|
| X | Cannot be specified | Cannot be specified | |
| 0 | No metastasis in regional lymph nodes | No distant metastasis | |
| 1 | <5 cm, no extra-adrenal invasion | With metastasis in regional lymph nodes | With distant metastasis |
| • M1a: to bone | |||
| • M1b: to non-regional lymph nodes, liver or lung. | |||
| • M1c: to bone and other organs | |||
| 2 | ≥5 cm, no extra-adrenal invasion or sympathetic EAPGL of any size | ||
| 3 | Any size, with extra-adrenal invasion |
Stage I: T1 N0 M0 (blank). Stage II: T2 N0 M0 (light grey). Stage III: T 1-2 N1 M0, T3 N0-1 M0 (mid grey). Stage IV: T 1-2 -3 N0-1 M1 (dark grey).
Adapted from The Eighth Edition AJCC Cancer Staging Manual. Springer, 2017.
The most common metastases occur in regional lymph nodes, bone, liver and lung.3,6–8
Pheochromocytoma and geneticsFrom 10% to 40% of patients have pheochromocytomas or EAPGL as part of a hereditary syndrome (Table 2). They tend to be younger, with smaller and more homogeneous lesions, more frequently bilateral.9–11 The following stand out: von Hippel-Lindau (Fig. 1); multiple endocrine neoplasia 2 (MEN 2) (Fig. 2); hereditary paraganglioma; and neurofibromatosis 1.
Pheochromocytomas and EAPGL in hereditary syndromes.
| Genes involved (and loci) | Type of inheritance | Epidemiology | % PCC and EAPGL | Approx. % malignancy | Other relevant data | Other conditions associated with the syndrome | |
|---|---|---|---|---|---|---|---|
| Von Hipple-Lindau | VHL (3p25-26) | AD, high penetrance | 1/36,000−50,000 live births | 25−30% PCC | <5% | Type 1. Low risk PCC | Cerebellar HB |
| PCC at mean age of 27 | 15% EAPGL | Type 2 A. High risk PCC and low ccRCC | Medullary HB | ||||
| 20−50% PCC bilateral | Type 2 B. High risk PCC and ccRCC | ccRCC | |||||
| Type 2 C. High risk PCC only | Renal cysts | ||||||
| Pancreatic NET | |||||||
| Pancreatic cysts | |||||||
| Pancreatic SCA | |||||||
| Tumour of the endolymphatic sac | |||||||
| Epididymal papillary cystadenomas | |||||||
| MEN 2 | RET (10q11.21) | AD, very high penetrance | Prevalence 1/30,000 | 40−50% PCC | 5% | MEN 2 A: classic forms, with cutaneous lichen amyloidosis and Hirchsprung disease, all with a similar incidence of PCC | MEN 2 A: |
| MEN 2 B: 6% of MEN 2 | EAPGL uncommon | Medullary thyroid cancer (>90%) | |||||
| 30−100% bilateral PCC | Parathyroid hyperplasia with hyperparathyroidism (10−20%) | ||||||
| MEN 2 B: | |||||||
| Medullary thyroid cancer (>90%, more aggressive) | |||||||
| Mucosal neuromas | |||||||
| Intestinal ganglioneuromas | |||||||
| Colonic aganglionosis | |||||||
| Marfanoid habitus | |||||||
| Myelination of corneal nerves. | |||||||
| Hereditary PGLa | PGL 1: SDHD (11q23). | AD, intermediate penetrance | Uncommon. Actual incidence unknown | PGL 1: PCC and EAPGL S 40%, PS EAPGL > 80% | PGL 1: <2% | Rather than the terms PGL 1−5, the genetic abnormality tends to be used instead (MAX, SDHA, SDHAF2, SDHB, SDHC, SDHD and TMEM127) | GIST (above all gastric) |
| PGL 4: SDHB (11p35) | 74% bilateral tumours | PGL 4: S EAPGL > 80% | PGL 4: 35−70% | Chondroma of the lung | |||
| ccRCC | |||||||
| Other | |||||||
| NF1 | NF1 (17q11.2) | AD, complete penetrance | 1/2,600−3,000 live births | 3% PCC | 12% | It is unusual to have EAPG OR bilateral PCC | Café-au-lait skin spots |
| 50% not inherited | Inguinal/axillary freckling | ||||||
| Lisch nodules of the iris | |||||||
| Cutaneous and neural neurofibromas | |||||||
| Plexiform and nodular neurofibromas | |||||||
| Optical and other gliomas | |||||||
| Soft tissue sarcoma | |||||||
| Malignant nerve sheath tumours | |||||||
| GIST | |||||||
| Glomus tumours | |||||||
| Other benign or malignant tumours | |||||||
| Bone dysplasias and pseudarthrosis | |||||||
| Other lesions bone | |||||||
| Cognitive deficit | |||||||
| Macrocephaly | |||||||
| Congenital heart disease | |||||||
| Hypertension | |||||||
| Other |
AD: autosomal dominant; SCA: serous cystadenomas; ccRCC: clear cell renal cell carcinomas; HB: haemangioblastomas; PCC: pheochromocytoma; GIST: gastrointestinal stromal tumour; MEN 2: multiple endocrine neoplasia type 2; NF1: neurofibromatosis type 1; EAPGL: extra-adrenal paraganglioma; PGL: paraganglioma; PS: parasympathetic; S: sympathetic; NET: neuroendocrine tumours.
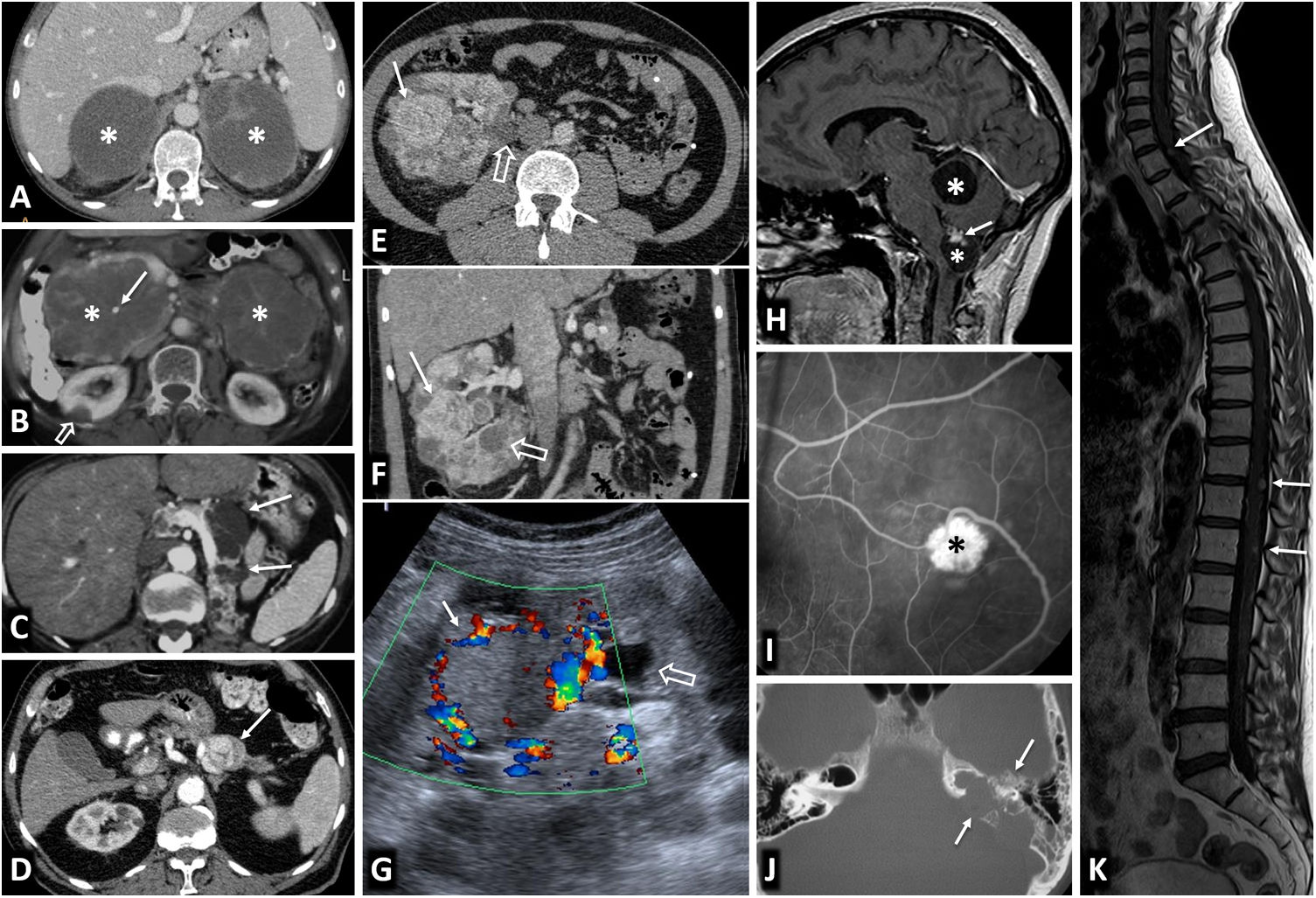
![Von Hippel-Lindau syndrome. Findings in different patients. A) Bilateral adrenal pheochromocytomas in a young male. CT: large well-defined masses in both adrenal glands (*), with predominant cystic areas. B) Multiple pancreatic serous cystadenomas in a young female. CT: two large multicystic pancreatic masses (*) can be seen, with radiating septa and, on the right, a small central calcification (arrow). A right cortical renal cyst can also be seen (hollow arrow). C) Multiple pancreatic cysts. CT: cysts of various sizes (arrows) in the body and tail of a repositioned pancreas after left nephrectomy for multiple clear cell renal carcinomas. D) Pancreatic neuroendocrine tumour. CT: small nodular lesion (arrow) with intense enhancement in the arterial phase, in the body of the pancreas. Small renal cysts can also be seen. E, F [CT], and G [Doppler ultrasound]) Multiple clear cell carcinomas (arrows) and cortical cysts (hollow arrows) in the single right kidney of a young male who has already had a left nephrectomy. H). Two cerebellar haemangioblastomas. Sagittal T1 MRI with Gd: cerebellar cystic lesions (*), with a solid nodule with heterogeneous enhancement in the lower one (arrow). I) Retinal angioma. Retinal fluorescein angiogram. Hyperfluorescent lesion (*) with a prominent arterial supply. J) Tumour of the endolymphatic sac. CT: lytic lesion (arrows) with its epicentre in the posterior petrous crest in the region of the vestibular aqueduct, with some intratumoral bone spicules. K) Multiple spinal haemangioblastomas. Sagittal, T1-weighted, Gd-enhanced MRI: small foci of intramedullary enhancement (arrows).](https://static.elsevier.es/multimedia/21735107/0000006400000004/v1_202208250603/S217351072200091X/v1_202208250603/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNfYM26a8Bai41R8kcJS5RRAf6ffZrG0r8IP2HfWtJ9XVXZ5z2r3pCrQW8lORcm8QNromXdu4rQQ4FaRKz0WspbNuYX3l184ECPG+HFB5GlIHVWXvwU57i21MGvx4dm6meURZSNbXDXXMS86SUnhipUDDu/CqDPxOm4UMUIjdB3cZ95rgZ7mOTfqBrSsNJCI6TLq7Fgv3wx6fxVGmDvFNBTTh0+jbfFjW3lx7ZZqEfujT9BXyNIyowXDySqjx4sa5QM= "Von Hippel-Lindau syndrome. Findings in different patients. A) Bilateral adrenal pheochromocytomas in a young male. CT: large well-defined masses in both adrenal glands (*), with predominant cystic areas. B) Multiple pancreatic serous cystadenomas in a young female. CT: two large multicystic pancreatic masses (*) can be seen, with radiating septa and, on the right, a small central calcification (arrow). A right cortical renal cyst can also be seen (hollow arrow). C) Multiple pancreatic cysts. CT: cysts of various sizes (arrows) in the body and tail of a repositioned pancreas after left nephrectomy for multiple clear cell renal carcinomas. D) Pancreatic neuroendocrine tumour. CT: small nodular lesion (arrow) with intense enhancement in the arterial phase, in the body of the pancreas. Small renal cysts can also be seen. E, F [CT], and G [Doppler ultrasound]) Multiple clear cell carcinomas (arrows) and cortical cysts (hollow arrows) in the single right kidney of a young male who has already had a left nephrectomy. H). Two cerebellar haemangioblastomas. Sagittal T1 MRI with Gd: cerebellar cystic lesions (*), with a solid nodule with heterogeneous enhancement in the lower one (arrow). I) Retinal angioma. Retinal fluorescein angiogram. Hyperfluorescent lesion (*) with a prominent arterial supply. J) Tumour of the endolymphatic sac. CT: lytic lesion (arrows) with its epicentre in the posterior petrous crest in the region of the vestibular aqueduct, with some intratumoral bone spicules. K) Multiple spinal haemangioblastomas. Sagittal, T1-weighted, Gd-enhanced MRI: small foci of intramedullary enhancement (arrows).")
Von Hippel-Lindau syndrome. Findings in different patients. A) Bilateral adrenal pheochromocytomas in a young male. CT: large well-defined masses in both adrenal glands (*), with predominant cystic areas. B) Multiple pancreatic serous cystadenomas in a young female. CT: two large multicystic pancreatic masses (*) can be seen, with radiating septa and, on the right, a small central calcification (arrow). A right cortical renal cyst can also be seen (hollow arrow). C) Multiple pancreatic cysts. CT: cysts of various sizes (arrows) in the body and tail of a repositioned pancreas after left nephrectomy for multiple clear cell renal carcinomas. D) Pancreatic neuroendocrine tumour. CT: small nodular lesion (arrow) with intense enhancement in the arterial phase, in the body of the pancreas. Small renal cysts can also be seen. E, F [CT], and G [Doppler ultrasound]) Multiple clear cell carcinomas (arrows) and cortical cysts (hollow arrows) in the single right kidney of a young male who has already had a left nephrectomy. H). Two cerebellar haemangioblastomas. Sagittal T1 MRI with Gd: cerebellar cystic lesions (*), with a solid nodule with heterogeneous enhancement in the lower one (arrow). I) Retinal angioma. Retinal fluorescein angiogram. Hyperfluorescent lesion (*) with a prominent arterial supply. J) Tumour of the endolymphatic sac. CT: lytic lesion (arrows) with its epicentre in the posterior petrous crest in the region of the vestibular aqueduct, with some intratumoral bone spicules. K) Multiple spinal haemangioblastomas. Sagittal, T1-weighted, Gd-enhanced MRI: small foci of intramedullary enhancement (arrows).
. 42-year-old female who consulted for episodes of hypoglycaemia. Left column, CT: two large and well-defined adrenal masses, with intensely enhancing solid areas(s), and other cystic areas (*) without significant enhancement. Two columns on the right, MRI: respective adrenal masses, with some foci of especially high signal on T2, corresponding to foci of cystic degeneration (*) and an area with high peripheral signal on T1, with very low signal on T2 (+), which corresponds to a focus of old haemorrhage with peripheral deposit of haemosiderin.")
MEN2A syndrome with bilateral pheochromocytoma and millimetre-sized medullary thyroid carcinoma (not shown, two foci identified in prophylactic total thyroidectomy). 42-year-old female who consulted for episodes of hypoglycaemia. Left column, CT: two large and well-defined adrenal masses, with intensely enhancing solid areas(s), and other cystic areas (*) without significant enhancement. Two columns on the right, MRI: respective adrenal masses, with some foci of especially high signal on T2, corresponding to foci of cystic degeneration (*) and an area with high peripheral signal on T1, with very low signal on T2 (+), which corresponds to a focus of old haemorrhage with peripheral deposit of haemosiderin.
In the region of 24%–70% of patients with pheochromocytoma-EAPGL considered sporadic (incidental or not) have some genetic abnormality (Table 3) in the pseudohypoxia lines, involving inappropriate activation of the hypoxia-inducing factor (clusters 1A -1B), kinase signalling, with stimulation of PI3K or RAS tumour pathways (cluster 2, more common in pheochromocytoma and associated with less aggressiveness) and Wnt signalling, involving DNA hypomethylation and poor prognosis (cluster 3), with many known and yet to be defined diagnostic and therapeutic consequences.8,10–13
Genetic abnormalities described in extra-adrenal pheochromocytomas and paragangliomas according to The Cancer Genome Atlas.
| Pathway involved | Estimated % | Genes | Type of tumour | Risk of metastasis % | Other syndromes and tumours | |
|---|---|---|---|---|---|---|
| Cluster 1A (VHL/EPAS1) | Pseudohypoxia (activation of hypoxia-inducing factor) | 15−20 | VHL | PCC>>EAPGL | 5 | VHL |
| EPAS1/HIF2α | PCC/TAPGL | 29 | PC, Somatostatinoma | |||
| EGLN1,2 | PCC/TAPGL | ?? | PC | |||
| Cluster 1B | 10−15 | SDHx | EAPGL | Low | GIST, RCC, PA | |
| (TCA) | SDHA | TAPGL>>H&NPGL/PCC | 30−70 | GIST, RCC, PA | ||
| SDHB | H&NPGL | Low | GIST, RCC | |||
| SDHC | EAPGL > PCC | <5 | GIST, RCC, PA | |||
| SDHD | H&NPGL | Low | Leiomyoma, RCC | |||
| SDHAF2 | PCC/EAPGL | >50 | Low-grade glioma | |||
| FH | TAPGL | ?? | AML | |||
| MDH2 | EAPGL | ?? | AML | |||
| IDH1/IDH2 | EAPGL | High? | ||||
| SLC25A11 | H&NPGL | ?? | ||||
| IDH3B | TAPGL | High? | ||||
| GOT2 | H&NPGL | ?? | ||||
| DNMT3A | PCC/EAPGL | ?? | ||||
| DLST | ||||||
| Cluster 2 | Kinase Signalling | 50−60 | RET | PCC | <5 | MEN 2 |
| NF1 | PCC | 12 | NF1 | |||
| TMEM 127 | PCC > EAPGL | Low | RCC | |||
| MAX | PCC/EAPGL | 10 | Renal oncocytoma | |||
| H-RAS | PCC | Low | Neuroblastoma | |||
| KIF1B | PCC | ?? | MEN 1 | |||
| MEN1 | PCC/H&NPGL | ?? | ||||
| Cluster 3 | Wnt Signalling | 5−10 | MAML3 | PCC/H&NPGL | High | Neuroblastoma |
| CSDE1 | PCC/H&NPGL | High |
A fourth cluster has recently been described, but is still poorly defined. In addition, relationships have been reported with other genes commonly involved in tumours, such as TP53 and BRAF.
PA: pituitary adenoma; RCC: renal cell carcinoma; PCC: pheochromocytoma; GIST: gastrointestinal stromal tumour; AML: acute myeloid leukaemia; MEN: multiple endocrine neoplasia; PC: polycythaemia; EAPGL: extra-adrenal paraganglioma; H&NPGL: head and neck paraganglioma; TAPGL: thoracic or abdominal paraganglioma; TCA: tricarboxylic acid; VHL: von Hippel-Lindau.
Adapted from Koopman et al.2.
Compatible symptoms, with or without hypertension, diagnosis of hypertension at an early age or an indicative family history are the main grounds for biochemical or genetic study. The distinctive diagnostic features can be consulted in guidelines and reference articles.3,6–8,14–16
After biochemical confirmation or where there is a high degree of clinical suspicion, CT or MRI should initially be performed, depending on the peculiarities of the case and the environment.
We have to remember that percutaneous puncture is contraindicated due to the risk of triggering a crisis relating to catecholamine discharge.
Nuclear medicine studies are used when the results of CT and MRI are negative, but where a high degree of suspicion persists or in uncertain cases.17–19 particularly the following:
- -
(123I) MIBG SPECT, with diagnostic accuracy of 95%, superior to scintigraphy (Fig. 3).
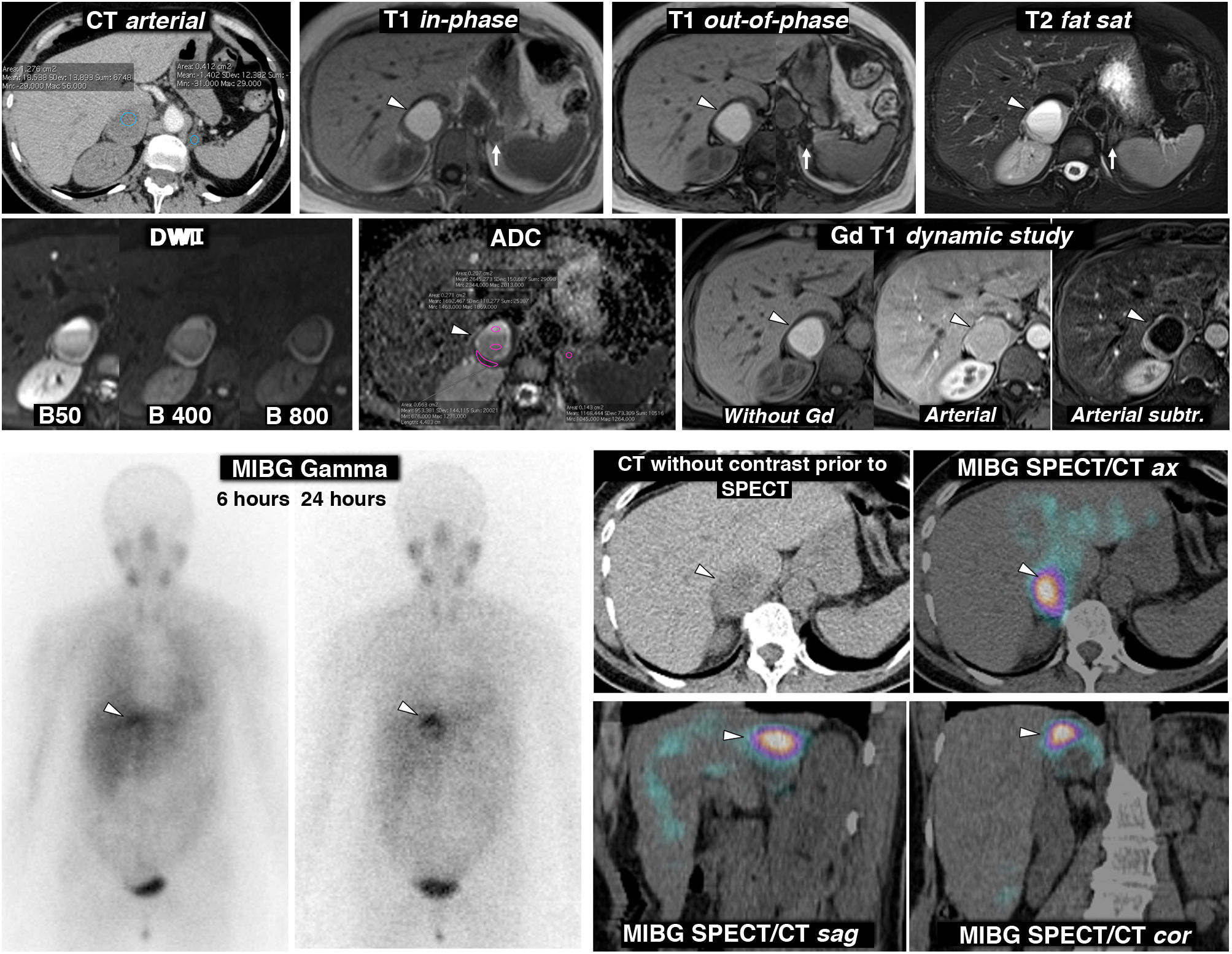
, a larger right one, with mean attenuation values somewhat lower than 20 HU, nonspecific, and a smaller left one of -1.4 HU, characterised as a lipid-rich adenoma. The MRI shows that the right lesion (arrowheads) is an almost purely haemorrhagic lesion, with a high signal on T1, without a drop in signal out-of-phase in the interior or in its periphery. The left adrenal adenoma (arrows) does lose signal intensely out-of-phase (a montage of images has been made to show the centre of the two lesions in the same image). The right adrenal lesion shows a high signal on T2, with two fluid levels, which can also be seen in the diffusion-weighted sequence and in the ADC map, with progressively greater restriction of diffusion towards gravitational areas. In the wall of this lesion, the mean ADC values are 0.95 × 10−3 mm2/s. The adenoma has mean values of 1.17. As in other haemorrhagic lesions, recognition of parietal or nodular enhancements is facilitated by subtracting the baseline image. Bottom: scintigraphy and SPET/CT findings with (123I) MIBG. The scintigraphy study shows uptake by the right lesion (arrowheads), more evident in the study 24 h after the administration of the radiopharmaceutical. The SPECT-CT study with the same radiopharmaceutical acquired at 6 h shows the uptake by the lesion more clearly, virtually confirming the diagnosis of pheochromocytoma. The left adenoma does not enhance.") Figure 3.
Figure 3.Pheochromocytoma as an almost purely haemorrhagic mass demonstrated by Nuclear Medicine. 58-year-old female. Went to the Accident and Emergency Department with headache and palpitations. Top: CT and MRI. Two adrenal nodules (elliptical ROI on CT), a larger right one, with mean attenuation values somewhat lower than 20 HU, nonspecific, and a smaller left one of -1.4 HU, characterised as a lipid-rich adenoma. The MRI shows that the right lesion (arrowheads) is an almost purely haemorrhagic lesion, with a high signal on T1, without a drop in signal out-of-phase in the interior or in its periphery. The left adrenal adenoma (arrows) does lose signal intensely out-of-phase (a montage of images has been made to show the centre of the two lesions in the same image). The right adrenal lesion shows a high signal on T2, with two fluid levels, which can also be seen in the diffusion-weighted sequence and in the ADC map, with progressively greater restriction of diffusion towards gravitational areas. In the wall of this lesion, the mean ADC values are 0.95 × 10−3 mm2/s. The adenoma has mean values of 1.17. As in other haemorrhagic lesions, recognition of parietal or nodular enhancements is facilitated by subtracting the baseline image. Bottom: scintigraphy and SPET/CT findings with (123I) MIBG. The scintigraphy study shows uptake by the right lesion (arrowheads), more evident in the study 24 h after the administration of the radiopharmaceutical. The SPECT-CT study with the same radiopharmaceutical acquired at 6 h shows the uptake by the lesion more clearly, virtually confirming the diagnosis of pheochromocytoma. The left adenoma does not enhance.
(0.42MB). - -
(111In) Octreotide SPECT, with more sensitivity in the detection of metastasis.
- -
(18F) FDG-PET/CT, very sensitive for detecting metastasis, with low specificity (Fig. 4).
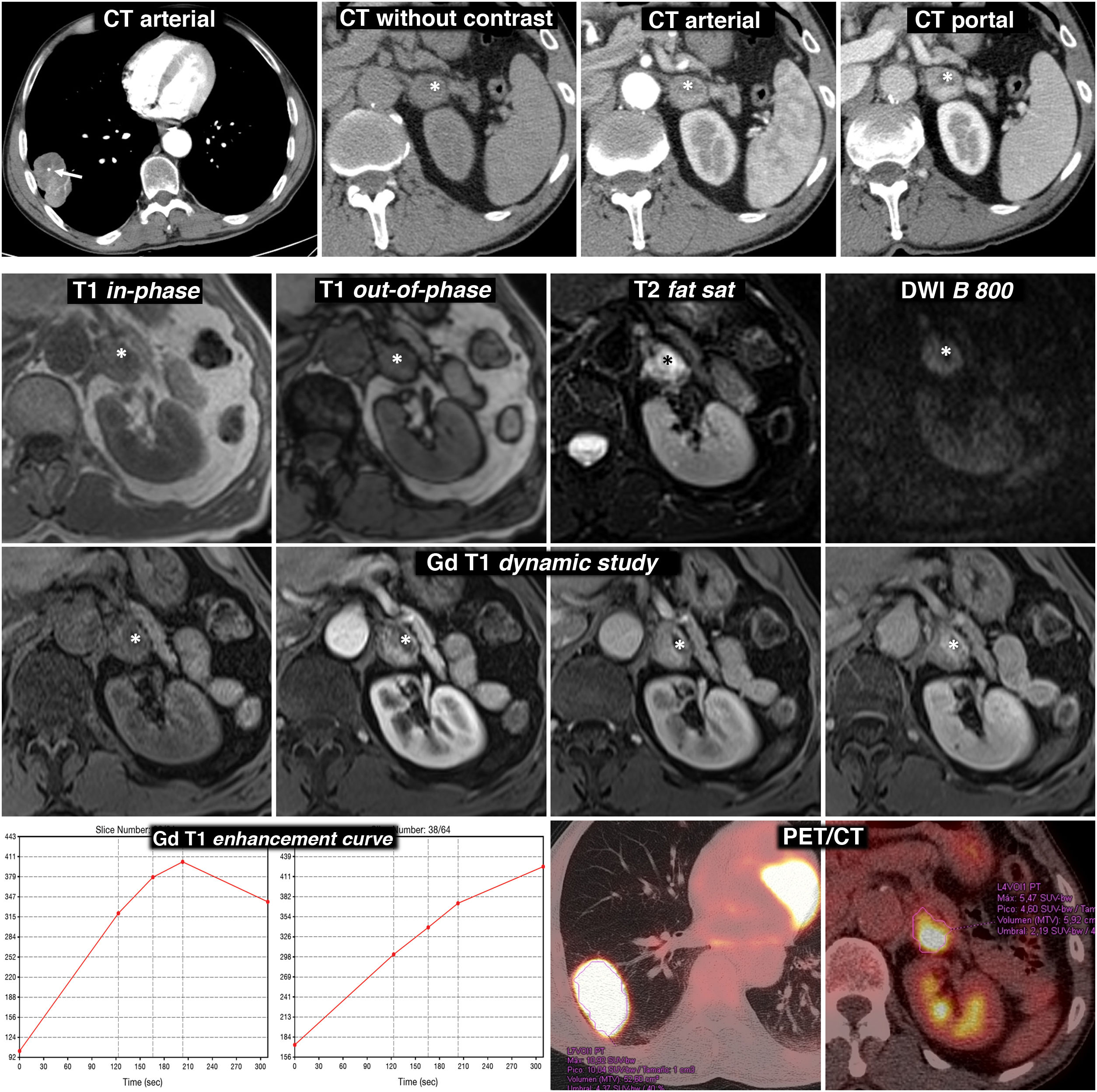
 and relatively intense and heterogeneous enhancement. Nodular lesion in the left adrenal gland with intense, rapid and heterogeneous enhancement. In the baseline phase of the study, there is an anterior area with attenuation values below 10 HU (*). Although interpreted as possibly corresponding to a cystic or necrotic focus rather than lipid content, as this region enhances less intensely, MRI is indicated. Bottom three rows, MR and PET/CT: the region with the lowest density on CT effectively corresponds to a cystic or necrotic focus (*): low signal on T1, very high on T2, no restriction of water diffusion and no significant enhancement. The solid areas of the lesion do not lose signal out-of-phase, have a high signal in T2, show moderate diffusion restriction and enhance intensely, some areas more quickly and others later, as can be seen in the enhancement curves. The suspected radiological diagnosis was metastasis. (18F) FDG PET/CT was performed, which showed intense metabolic activity both in the lung lesion and in the adrenal gland. The histological study of the lung lesion showed adenocarcinoma of the lung and that of the adrenal gland, pheochromocytoma. There were no surgical complications despite the fact that no pharmacological preparation was performed.") Figure 4.
Figure 4.Diagnostic error. Adrenal pheochromocytoma interpreted as lung adenocarcinoma metastasis. 69-year-old male smoker. Finding of lung mass on chest X-ray. Top row, CT: lobulated and well-defined lung tumour mass with punctate calcification (arrow) and relatively intense and heterogeneous enhancement. Nodular lesion in the left adrenal gland with intense, rapid and heterogeneous enhancement. In the baseline phase of the study, there is an anterior area with attenuation values below 10 HU (*). Although interpreted as possibly corresponding to a cystic or necrotic focus rather than lipid content, as this region enhances less intensely, MRI is indicated. Bottom three rows, MR and PET/CT: the region with the lowest density on CT effectively corresponds to a cystic or necrotic focus (*): low signal on T1, very high on T2, no restriction of water diffusion and no significant enhancement. The solid areas of the lesion do not lose signal out-of-phase, have a high signal in T2, show moderate diffusion restriction and enhance intensely, some areas more quickly and others later, as can be seen in the enhancement curves. The suspected radiological diagnosis was metastasis. (18F) FDG PET/CT was performed, which showed intense metabolic activity both in the lung lesion and in the adrenal gland. The histological study of the lung lesion showed adenocarcinoma of the lung and that of the adrenal gland, pheochromocytoma. There were no surgical complications despite the fact that no pharmacological preparation was performed.
(0.73MB). - -
(68Ga) DOTATATE PET/CT, occasionally more sensitive for detecting metastatic disease, and with higher spatial resolution.
, a larger right one, with mean attenuation values somewhat lower than 20 HU, nonspecific, and a smaller left one of -1.4 HU, characterised as a lipid-rich adenoma. The MRI shows that the right lesion (arrowheads) is an almost purely haemorrhagic lesion, with a high signal on T1, without a drop in signal out-of-phase in the interior or in its periphery. The left adrenal adenoma (arrows) does lose signal intensely out-of-phase (a montage of images has been made to show the centre of the two lesions in the same image). The right adrenal lesion shows a high signal on T2, with two fluid levels, which can also be seen in the diffusion-weighted sequence and in the ADC map, with progressively greater restriction of diffusion towards gravitational areas. In the wall of this lesion, the mean ADC values are 0.95 × 10−3 mm2/s. The adenoma has mean values of 1.17. As in other haemorrhagic lesions, recognition of parietal or nodular enhancements is facilitated by subtracting the baseline image. Bottom: scintigraphy and SPET/CT findings with (123I) MIBG. The scintigraphy study shows uptake by the right lesion (arrowheads), more evident in the study 24 h after the administration of the radiopharmaceutical. The SPECT-CT study with the same radiopharmaceutical acquired at 6 h shows the uptake by the lesion more clearly, virtually confirming the diagnosis of pheochromocytoma. The left adenoma does not enhance.")
 and relatively intense and heterogeneous enhancement. Nodular lesion in the left adrenal gland with intense, rapid and heterogeneous enhancement. In the baseline phase of the study, there is an anterior area with attenuation values below 10 HU (*). Although interpreted as possibly corresponding to a cystic or necrotic focus rather than lipid content, as this region enhances less intensely, MRI is indicated. Bottom three rows, MR and PET/CT: the region with the lowest density on CT effectively corresponds to a cystic or necrotic focus (*): low signal on T1, very high on T2, no restriction of water diffusion and no significant enhancement. The solid areas of the lesion do not lose signal out-of-phase, have a high signal in T2, show moderate diffusion restriction and enhance intensely, some areas more quickly and others later, as can be seen in the enhancement curves. The suspected radiological diagnosis was metastasis. (18F) FDG PET/CT was performed, which showed intense metabolic activity both in the lung lesion and in the adrenal gland. The histological study of the lung lesion showed adenocarcinoma of the lung and that of the adrenal gland, pheochromocytoma. There were no surgical complications despite the fact that no pharmacological preparation was performed.")
There is currently no indication for selective hormonal sampling in adrenal veins.
Radiological findingsThey can be detected with ultrasound (with different degrees of echogenicity in the solid component), CT or MRI. Tables 4 and 5 respectively show our CT and MRI technical protocols for the study of adrenal nodules. The findings are non-specific, with the following data being indicative:20–26
- -
Good delimitation, even with large tumours (Figs. 1–10). Infiltrative behaviour has been correlated with greater biological aggressiveness.
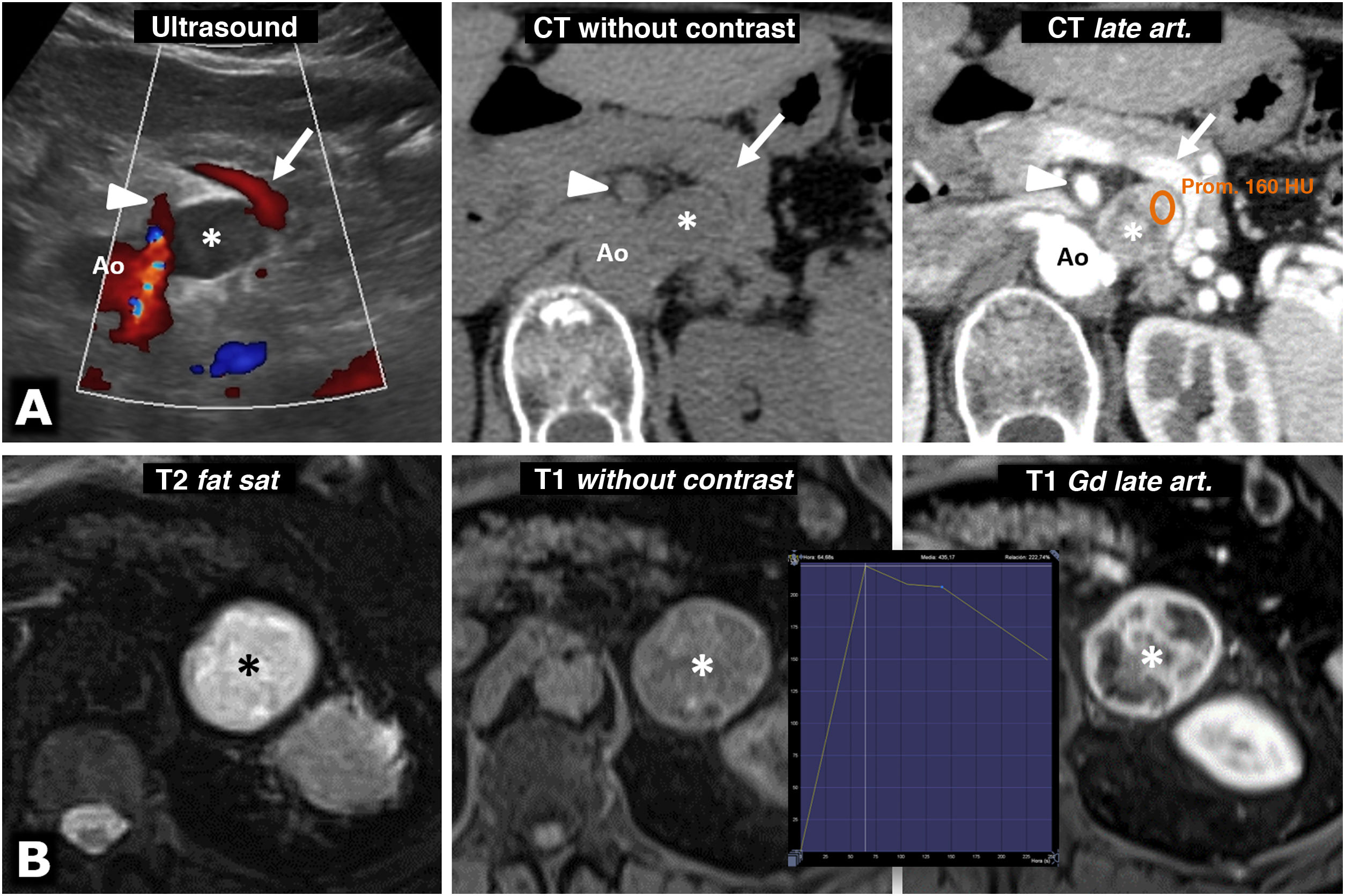
 74-year-old female. Incidental ultrasound finding of a well-defined, slightly heterogeneous solid hypoechoic nodule (*), located between the splenic vein (arrows), the superior mesenteric artery (arrowheads) and the aorta (Ao). The CT shows intense and heterogeneous enhancement in the late arterial phase, in which it reaches 160 HU in some areas, with moderate washout in later phases (not shown). B) 73-year-old male. During investigations for grade II hypertension, a left adrenal mass was found (*), which on MRI behaves with high signal intensity on T2-weighted images, with marked enhancement (relatively greater than 220%), with a peak in the late arterial phase of the dynamic study. The hypertension was corrected after laparoscopic adrenalectomy.") Figure 5.
Figure 5.Intense enhancement in pheochromocytomas. A) 74-year-old female. Incidental ultrasound finding of a well-defined, slightly heterogeneous solid hypoechoic nodule (*), located between the splenic vein (arrows), the superior mesenteric artery (arrowheads) and the aorta (Ao). The CT shows intense and heterogeneous enhancement in the late arterial phase, in which it reaches 160 HU in some areas, with moderate washout in later phases (not shown). B) 73-year-old male. During investigations for grade II hypertension, a left adrenal mass was found (*), which on MRI behaves with high signal intensity on T2-weighted images, with marked enhancement (relatively greater than 220%), with a peak in the late arterial phase of the dynamic study. The hypertension was corrected after laparoscopic adrenalectomy.
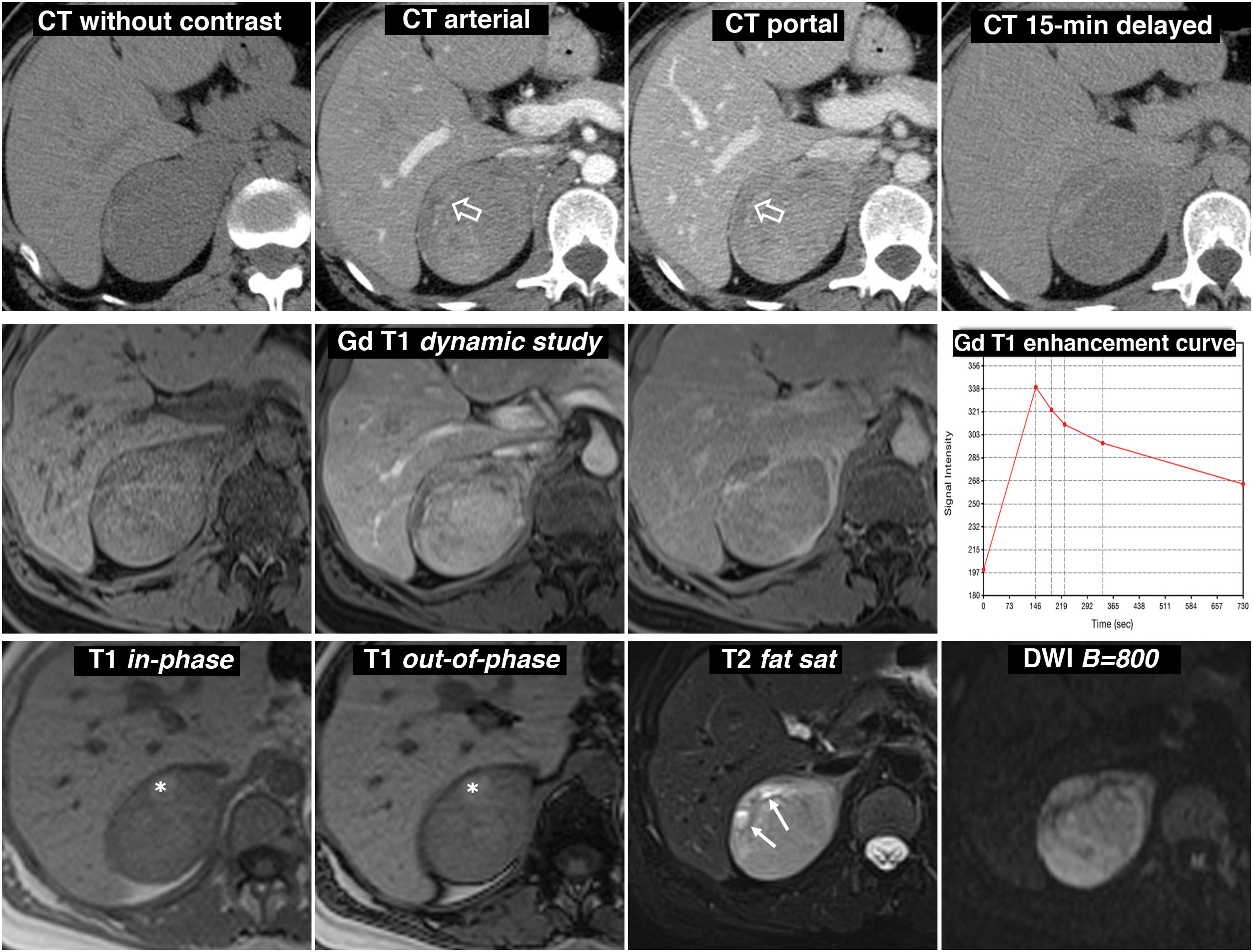
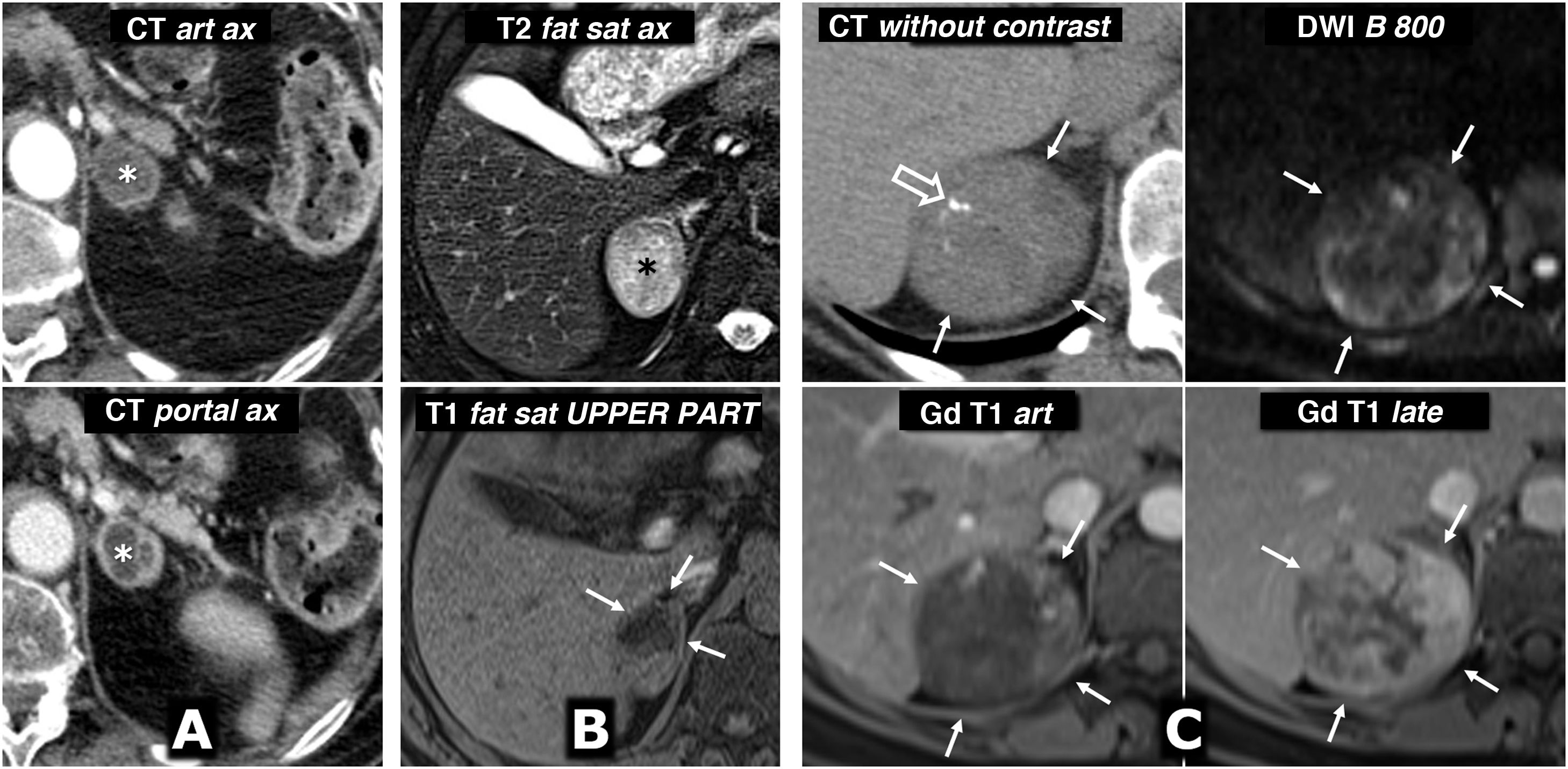
(0.49MB).. The mean attenuation values in the different phases of the study are 30 (does not show the presence of lipids), 63, 89 and 49 HU respectively, with absolute washout of 67.8% and relative washout of 44.9% in the late study, above 60% and 40% respectively, which is suggestive of adenoma with high reliability. In addition, some studies have indicated that the presence of vessels within a well-defined adrenal nodular lesion is highly indicative of adenoma. However, as one-third of pheochromocytomas can also have washout rates similar to adenomas, it was considered a nonspecific lesion and MRI was performed. Central row: dynamic study and enhancement curve in MRI with extracellular contrast. The mass shows intense enhancement (relative greater than 330%), with a peak in the arterial phase and subsequent washout. Bottom row: other MRI sequences. Does not show signal drop out-of-phase, shows high signal in T2, preserved in diffusion with high B and contains the odd small cystic (arrows) and haemorrhagic focus (*). The presence of cystic and haemorrhagic foci in a well-defined adrenal mass with the rest of the described characteristics led to a presumed diagnosis of pheochromocytoma, which was supported by elevated catecholamines in urine and later confirmed in the histological study of the surgical specimen.") Figure 6.
Figure 6.Predominantly solid pheochromocytoma, with late washout similar to the adenoma in the CT study. A 51-year-old female with a right adrenal mass found on PET ultrasound. Did not have hypertension. Top row, CT: well-defined and fairly homogeneous right adrenal mass with a vessel inside (hollow arrows). The mean attenuation values in the different phases of the study are 30 (does not show the presence of lipids), 63, 89 and 49 HU respectively, with absolute washout of 67.8% and relative washout of 44.9% in the late study, above 60% and 40% respectively, which is suggestive of adenoma with high reliability. In addition, some studies have indicated that the presence of vessels within a well-defined adrenal nodular lesion is highly indicative of adenoma. However, as one-third of pheochromocytomas can also have washout rates similar to adenomas, it was considered a nonspecific lesion and MRI was performed. Central row: dynamic study and enhancement curve in MRI with extracellular contrast. The mass shows intense enhancement (relative greater than 330%), with a peak in the arterial phase and subsequent washout. Bottom row: other MRI sequences. Does not show signal drop out-of-phase, shows high signal in T2, preserved in diffusion with high B and contains the odd small cystic (arrows) and haemorrhagic focus (*). The presence of cystic and haemorrhagic foci in a well-defined adrenal mass with the rest of the described characteristics led to a presumed diagnosis of pheochromocytoma, which was supported by elevated catecholamines in urine and later confirmed in the histological study of the surgical specimen.
(0.5MB). and heterogeneous echogenicity in the solid areas, with some areas more echogenic and others hypoechoic. Pheochromocytoma was suggested in the ultrasound diagnosis. The appearance on MR is also characteristic, with predominant solid areas with high signal on T2-weighted images (compare with that of the paravertebral musculature), which enhance intensely and do not show signal drop out-of-phase. There is a correlation between the most hypoechoic areas and the areas with greater restriction of water diffusion, which maintain a higher signal in the diffusion-weighted sequence with high B value and are markedly hypointense in the parametric ADC map (with values of 0.7 × 10−3 mm2/s). The cystic areas are hypointense on T1, hyperintense on T2, do not enhance and show facilitated water diffusion, with a large drop in signal with high B value and ADC of 2.2 × 10−3 mm2/s.") Figure 7.
Figure 7.Pheochromocytoma with cystic changes and high signal on T2. Ultrasound and MRI 30-year-old woman with severe headache who required admission to the ICU for a hypertensive emergency. Ultrasound revealed a practically spherical right adrenal mass, perfectly delimited, with small cystic foci (*) and heterogeneous echogenicity in the solid areas, with some areas more echogenic and others hypoechoic. Pheochromocytoma was suggested in the ultrasound diagnosis. The appearance on MR is also characteristic, with predominant solid areas with high signal on T2-weighted images (compare with that of the paravertebral musculature), which enhance intensely and do not show signal drop out-of-phase. There is a correlation between the most hypoechoic areas and the areas with greater restriction of water diffusion, which maintain a higher signal in the diffusion-weighted sequence with high B value and are markedly hypointense in the parametric ADC map (with values of 0.7 × 10−3 mm2/s). The cystic areas are hypointense on T1, hyperintense on T2, do not enhance and show facilitated water diffusion, with a large drop in signal with high B value and ADC of 2.2 × 10−3 mm2/s.
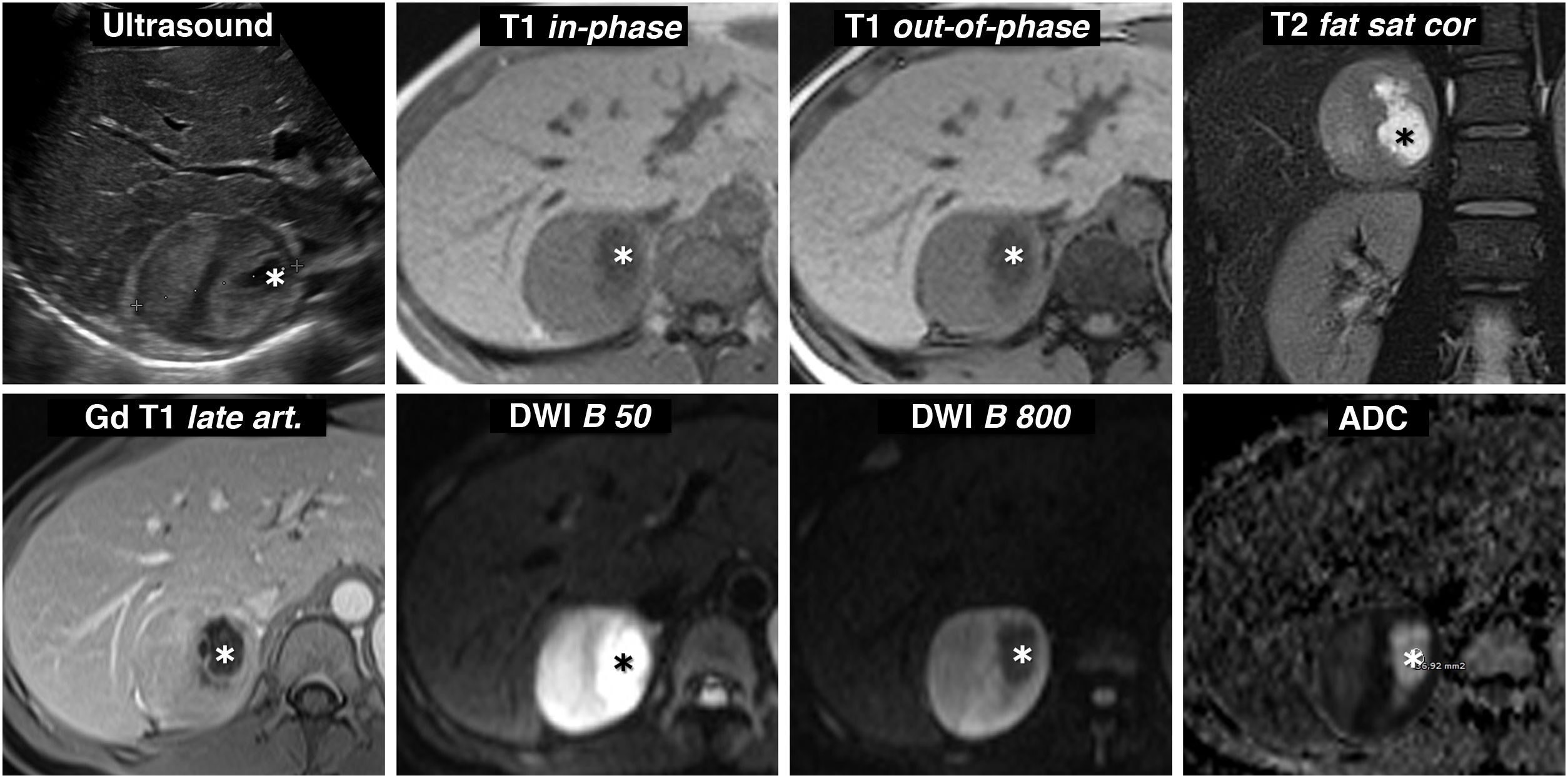
(0.28MB)., which show high signal on the ADC map due to the absence of restriction of water diffusion. These foci therefore lose the signal in the diffusion-weighted sequence with a high B value. In the dynamic study, with contrast, the periphery of the lesion shows intense enhancement, with the peak in the arterial phase of the study, while the centre, with cystic phenomena demonstrated by pathology examination, has little enhancement.") Figure 8.
Figure 8.Incidental finding of adrenal pheochromocytoma with cystic areas while being investigated due to abdominal trauma. 57-year-old male. Portal phase CT incidentally identifies a right adrenal nodular lesion with a more hypoattenuating centre and a hyperattenuating periphery, probably due to contrast enhancement. On the MRI, there is no signal drop out-of-phase. It is markedly hyperintense on T2-weighted images, with some cystic-like foci (arrows), which show high signal on the ADC map due to the absence of restriction of water diffusion. These foci therefore lose the signal in the diffusion-weighted sequence with a high B value. In the dynamic study, with contrast, the periphery of the lesion shows intense enhancement, with the peak in the arterial phase of the study, while the centre, with cystic phenomena demonstrated by pathology examination, has little enhancement.
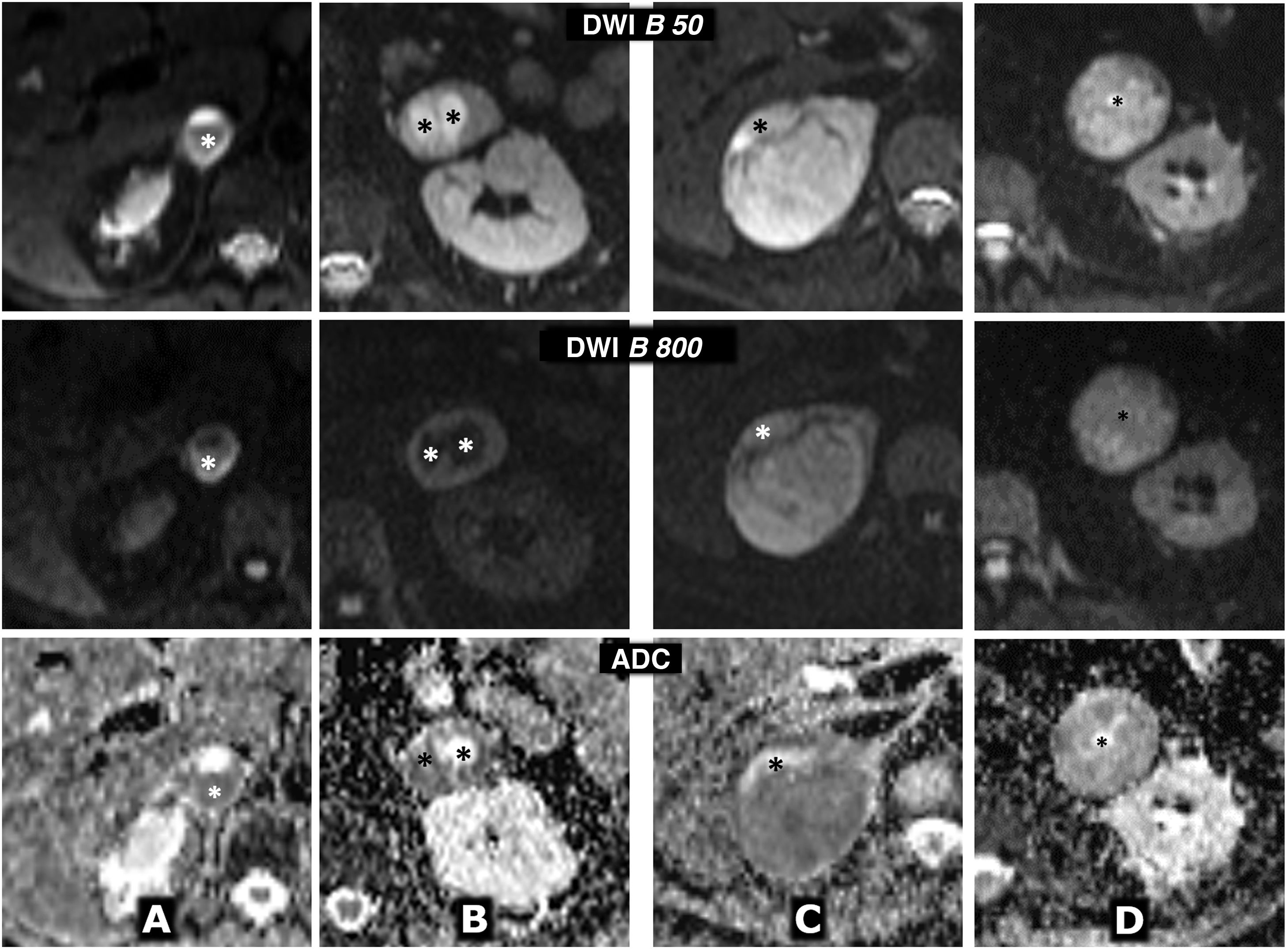
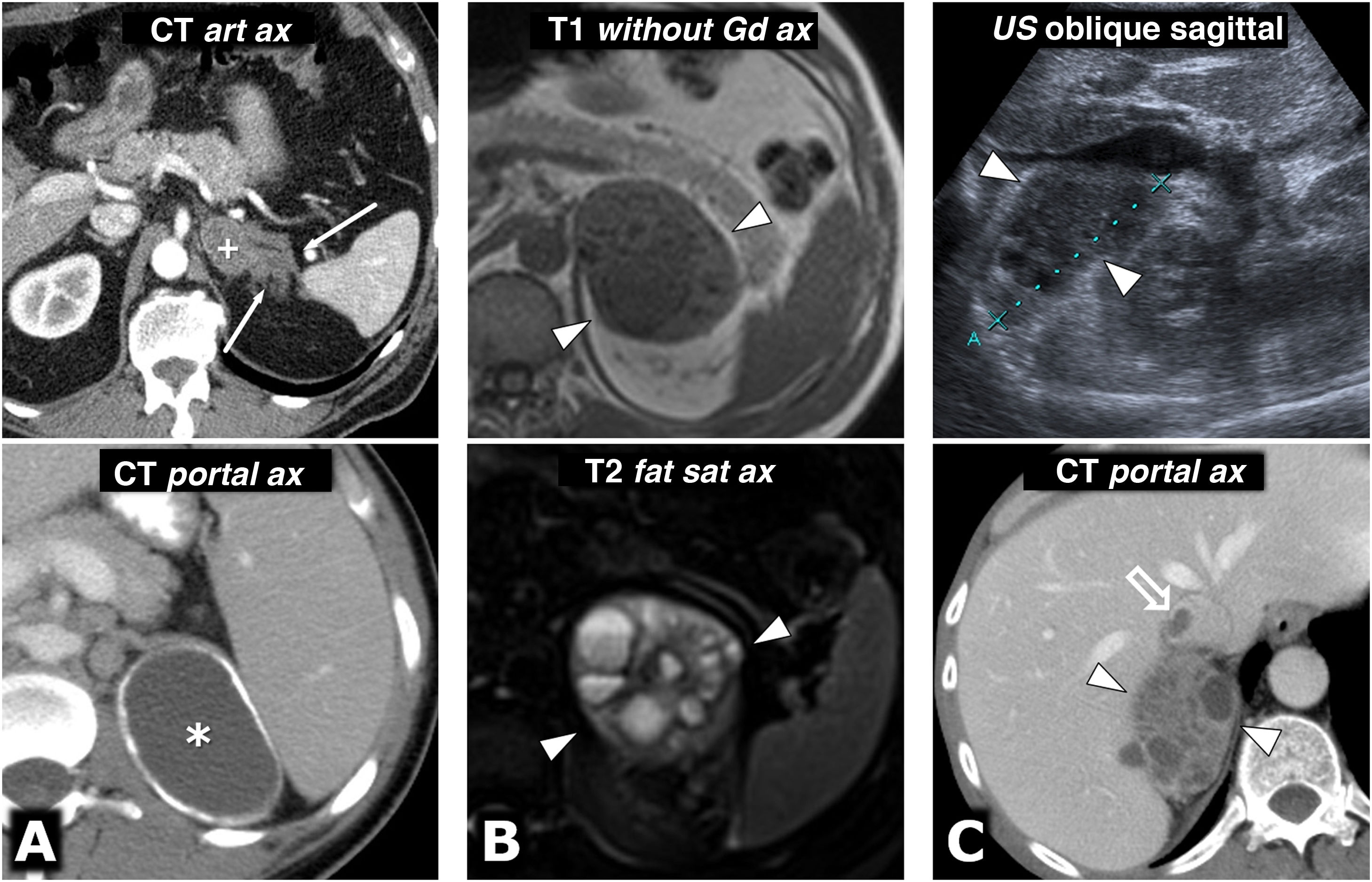
(0.31MB).. Centre row: diffusion sequence with B = 800 s/mm2 (enhanced in diffusion). Bottom row: apparent diffusion coefficient (ADC) map. A) 64-year-old female with hypertensive crises and paroxysms of headache. Right adrenal pheochromocytoma 22 mm in size with almost global cystic-haemorrhagic features, histologically demonstrated, with an antigravitational area with facilitated water diffusion and another gravitational (*) with low signal on T2 and T1 (not shown), relatively high in diffusion with B = 800 s/mm2 and low in the ADC map, with an average value of 0.9 × 10−3 mm2/s. The solid tissue wall, about 3 mm thick, shows high signal on T2 and diffusion restriction, with high signal at B = 800 and average ADC of 0.83. Ki-67 index in the surgical specimen: 2%. Has had no recurrence or progression 20 months later. B) A 49-year-old male with an incidental diagnosis of a left pheochromocytoma 43 mm in size confirmed by elevated serum and urine catecholamines and then histologically after laparoscopic resection. Predominantly solid mass, with two cystic areas (*) where water diffuses freely. In peripheral solid areas there is diffusion restriction, with an average ADC of 0.78 × 103 mm2/s. Ki-67 index: <1%. No recurrence or progression seven years later. C) 51-year-old female with hypertensive crises. Practically solid right pheochromocytoma 82 mm in size, with small anterior cystic foci (*) and facilitated diffusion of water. In solid areas there is a very high signal on T2 and moderate restriction of water diffusion, with an average ADC value of 0.94 × 10−3 mm2/s. Ki-67 index: 3%. No recurrence or progression six years later. D) 73-year-old male with grade II hypertension. Single left pheochromocytoma, practically solid, 51 mm in size, with very small central foci of cystic degeneration. Solid areas show high signal on T2 and relatively little restriction of water diffusion, with ADC of 1.3 × 10−3 mm2/s. Ki-67 index: 2%. No recurrence or progression a year after resection.") Figure 9.
Figure 9.Variable behaviour in diffusion in four different cases of pheochromocytoma. Top row: diffusion sequence with B = 50 s/mm2 (almost completely enhanced on T2). Centre row: diffusion sequence with B = 800 s/mm2 (enhanced in diffusion). Bottom row: apparent diffusion coefficient (ADC) map. A) 64-year-old female with hypertensive crises and paroxysms of headache. Right adrenal pheochromocytoma 22 mm in size with almost global cystic-haemorrhagic features, histologically demonstrated, with an antigravitational area with facilitated water diffusion and another gravitational (*) with low signal on T2 and T1 (not shown), relatively high in diffusion with B = 800 s/mm2 and low in the ADC map, with an average value of 0.9 × 10−3 mm2/s. The solid tissue wall, about 3 mm thick, shows high signal on T2 and diffusion restriction, with high signal at B = 800 and average ADC of 0.83. Ki-67 index in the surgical specimen: 2%. Has had no recurrence or progression 20 months later. B) A 49-year-old male with an incidental diagnosis of a left pheochromocytoma 43 mm in size confirmed by elevated serum and urine catecholamines and then histologically after laparoscopic resection. Predominantly solid mass, with two cystic areas (*) where water diffuses freely. In peripheral solid areas there is diffusion restriction, with an average ADC of 0.78 × 103 mm2/s. Ki-67 index: <1%. No recurrence or progression seven years later. C) 51-year-old female with hypertensive crises. Practically solid right pheochromocytoma 82 mm in size, with small anterior cystic foci (*) and facilitated diffusion of water. In solid areas there is a very high signal on T2 and moderate restriction of water diffusion, with an average ADC value of 0.94 × 10−3 mm2/s. Ki-67 index: 3%. No recurrence or progression six years later. D) 73-year-old male with grade II hypertension. Single left pheochromocytoma, practically solid, 51 mm in size, with very small central foci of cystic degeneration. Solid areas show high signal on T2 and relatively little restriction of water diffusion, with ADC of 1.3 × 10−3 mm2/s. Ki-67 index: 2%. No recurrence or progression a year after resection.
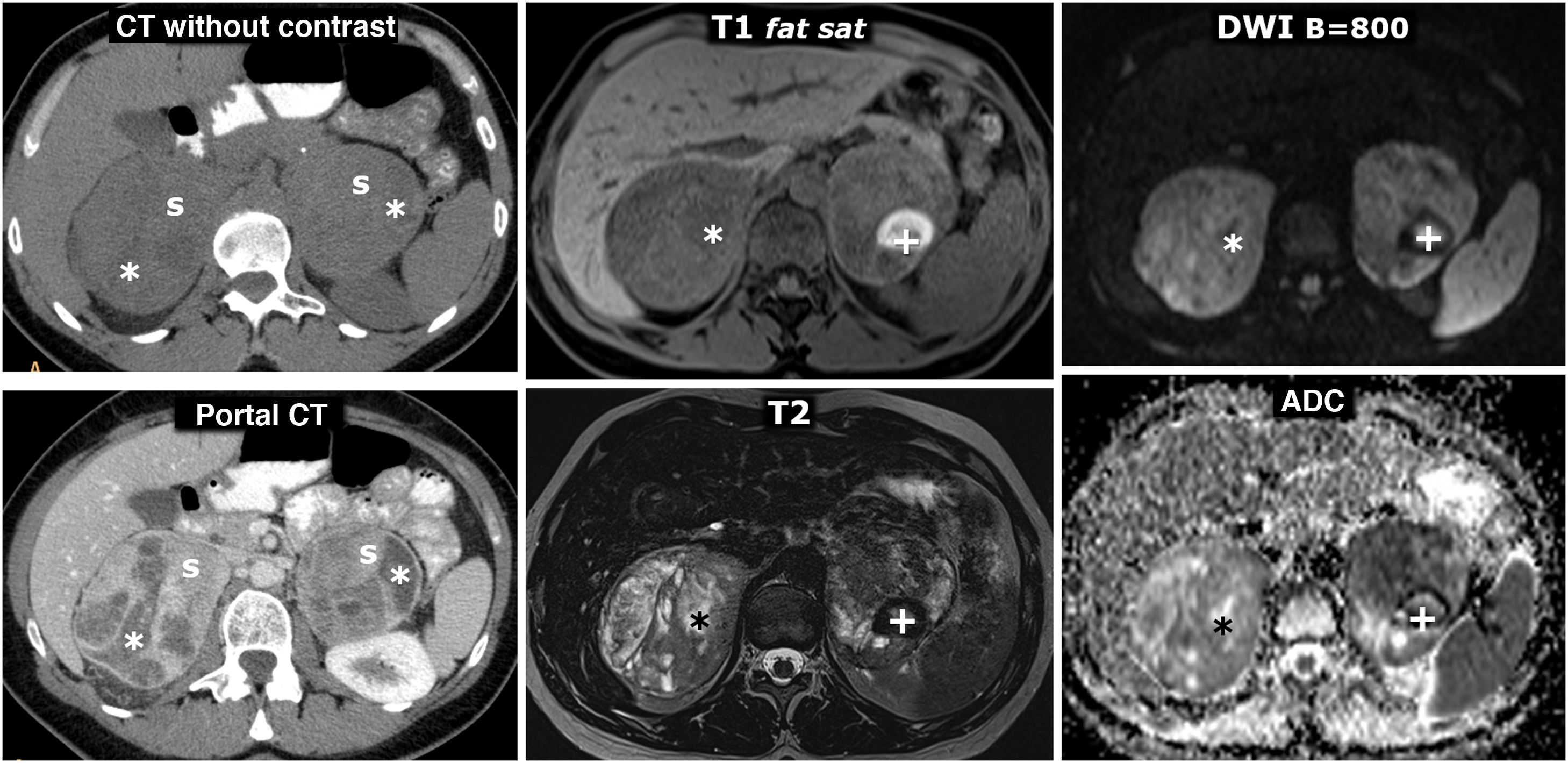
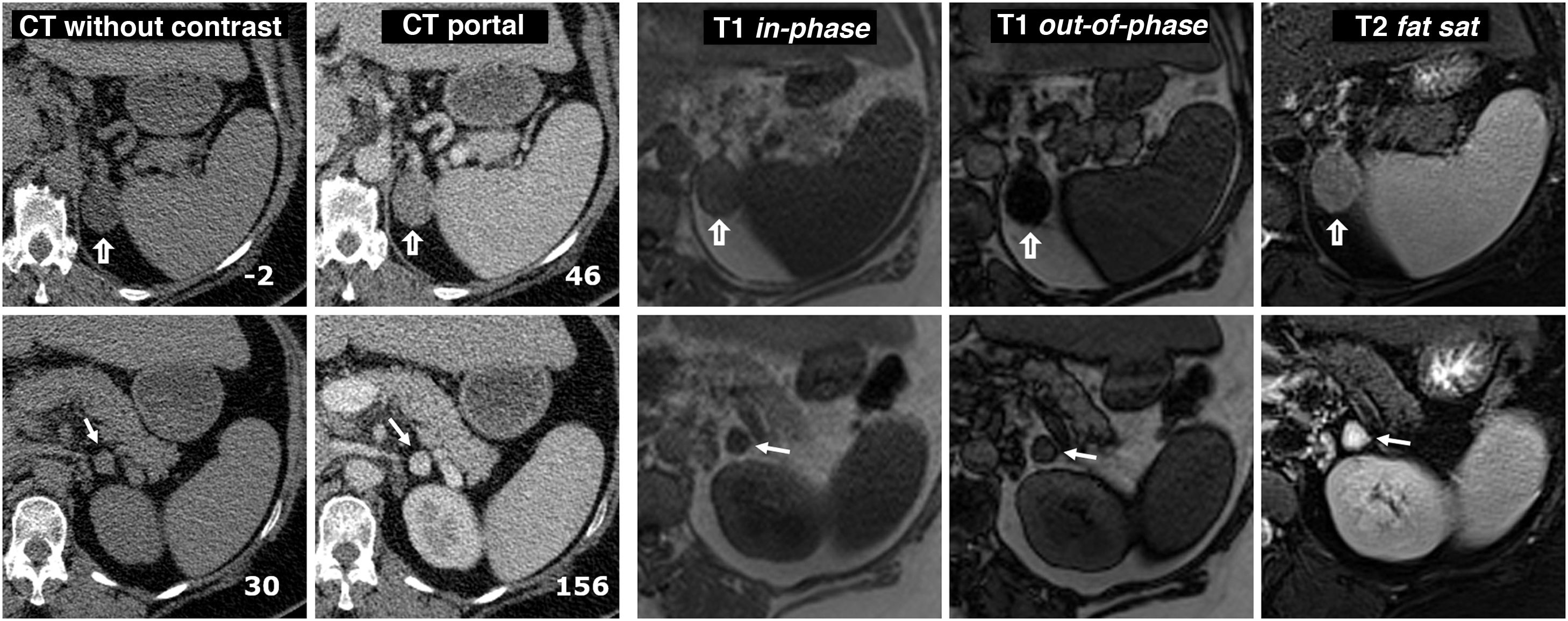
(0.5MB). are shown in the top row and the pheochromocytoma (arrows) in the bottom row, both confirmed histologically. Two left-hand columns, CT: the number at the bottom right of each box corresponds to the mean attenuation value of the lesion in the axial slice shown. The adenoma has negative baseline attenuation, which allows it to be characterised. That of the pheochromocytoma is 30 HU, which is nonspecific. It enhances intensely in the arterial phase, heterogeneously (not shown), and in the portal phase, more homogeneously, exceeding 130 HU. No late washout study was performed. Remaining columns on the right, MRI: the adenoma shows a large drop in signal out-of-phase, which reveals its microscopic lipid content. The pheochromocytoma does not show a significant drop in signal and is surrounded by “India ink” artifact. The quantitative study also does not show a significant lipid content. The adenoma has an intermediate signal on T2. The pheochromocytoma shows a very high signal (\"light bulb sign\").") Figure 10.
Figure 10.Collision tumour: adenoma and pheochromocytoma in the same adrenal gland. 58-year-old male. Incidental finding of two nodules in the left adrenal gland. Images of the adenoma (hollow arrows) are shown in the top row and the pheochromocytoma (arrows) in the bottom row, both confirmed histologically. Two left-hand columns, CT: the number at the bottom right of each box corresponds to the mean attenuation value of the lesion in the axial slice shown. The adenoma has negative baseline attenuation, which allows it to be characterised. That of the pheochromocytoma is 30 HU, which is nonspecific. It enhances intensely in the arterial phase, heterogeneously (not shown), and in the portal phase, more homogeneously, exceeding 130 HU. No late washout study was performed. Remaining columns on the right, MRI: the adenoma shows a large drop in signal out-of-phase, which reveals its microscopic lipid content. The pheochromocytoma does not show a significant drop in signal and is surrounded by “India ink” artifact. The quantitative study also does not show a significant lipid content. The adenoma has an intermediate signal on T2. The pheochromocytoma shows a very high signal ("light bulb sign").
(0.42MB). - -
Intense and rapid enhancement with iodinated contrast (they usually reach over 130 HU); or paramagnetic, not contraindicated (Fig. 5). May overlap with that of the adenoma.
- -
Late washout less than that of the adenoma, although in 35% of cases the values overlap25,26 (Fig. 6). In the remaining 65%, it cannot be differentiated from carcinoma and metastasis.
- -
High signal in T2-weighted sequence (“light bulb sign”) in solid tumour areas, with high specificity, but low sensitivity (Figs. 5 and 7). The measurement of the signal-to-noise ratio in T2-weighted sequence with respect to the paravertebral musculature could be more useful than qualitative assessment.25–27
- -
Frequent cystic and haemorrhagic changes (Figs. 1–3, 7 and 8), which makes them heterogeneous, particularly when they are large. The content may be hyperintense on T1-weighted sequence due to haemorrhage (Fig. 3). They may have a purely cystic appearance or show only a peripheral ring of solid tumour, with enhancement and behaviour on T2-weighted sequence as described. Sometimes the assessment of enhancement may require subtraction of the baseline image or spectral quantification.
- -
Absence of macro or microscopic lipids. Solid areas have attenuation values greater than 10 HU on CT without contrast (Figs. 2, 4 and 6), but they can be lower in cystic regions. The presence of macroscopic foci of fat in a pheochromocytoma is exceptional (less than 0.5% of cases). The absence of a significant drop in signal out-of-phase (Figs. 3, 4, 6, 7 and 8) is one of the data enabling a safer differential diagnosis with adenoma.25 Areas with high signal in T1-weighted sequence in MRI, common due to haemorrhagic phenomena, are not suppressed with fat saturation.
- -
The behaviour in diffusion-weighted sequences does not provide great diagnostic value. Cystic or necrotic areas generally show free diffusion, with high values of apparent diffusion coefficient (ADC), and solid areas tend to have restricted diffusion to a greater or lesser degree (Fig. 9). In general, there is overlap in ADC values for pheochromocytoma and other adrenal tumours, including adenomas. The exceptions are primary adrenal carcinomas and lymphomas, which exhibit marked diffusion restriction. Some studies have documented higher ADC values than in adenomas or metastases, ADC differences between benign and malignant, or histogram abnormalities, but these findings have not been confirmed.
- -
They may present calcifications (up to 10% in the literature, although very uncommon in our experience), generally punctate.
- -
The coexistence of a pheochromocytoma and another tumour in the same adrenal gland (collision tumour) is highly unusual, with the least uncommon being adenoma (Fig. 10).
 74-year-old female. Incidental ultrasound finding of a well-defined, slightly heterogeneous solid hypoechoic nodule (*), located between the splenic vein (arrows), the superior mesenteric artery (arrowheads) and the aorta (Ao). The CT shows intense and heterogeneous enhancement in the late arterial phase, in which it reaches 160 HU in some areas, with moderate washout in later phases (not shown). B) 73-year-old male. During investigations for grade II hypertension, a left adrenal mass was found (*), which on MRI behaves with high signal intensity on T2-weighted images, with marked enhancement (relatively greater than 220%), with a peak in the late arterial phase of the dynamic study. The hypertension was corrected after laparoscopic adrenalectomy.")
. The mean attenuation values in the different phases of the study are 30 (does not show the presence of lipids), 63, 89 and 49 HU respectively, with absolute washout of 67.8% and relative washout of 44.9% in the late study, above 60% and 40% respectively, which is suggestive of adenoma with high reliability. In addition, some studies have indicated that the presence of vessels within a well-defined adrenal nodular lesion is highly indicative of adenoma. However, as one-third of pheochromocytomas can also have washout rates similar to adenomas, it was considered a nonspecific lesion and MRI was performed. Central row: dynamic study and enhancement curve in MRI with extracellular contrast. The mass shows intense enhancement (relative greater than 330%), with a peak in the arterial phase and subsequent washout. Bottom row: other MRI sequences. Does not show signal drop out-of-phase, shows high signal in T2, preserved in diffusion with high B and contains the odd small cystic (arrows) and haemorrhagic focus (*). The presence of cystic and haemorrhagic foci in a well-defined adrenal mass with the rest of the described characteristics led to a presumed diagnosis of pheochromocytoma, which was supported by elevated catecholamines in urine and later confirmed in the histological study of the surgical specimen.")
 and heterogeneous echogenicity in the solid areas, with some areas more echogenic and others hypoechoic. Pheochromocytoma was suggested in the ultrasound diagnosis. The appearance on MR is also characteristic, with predominant solid areas with high signal on T2-weighted images (compare with that of the paravertebral musculature), which enhance intensely and do not show signal drop out-of-phase. There is a correlation between the most hypoechoic areas and the areas with greater restriction of water diffusion, which maintain a higher signal in the diffusion-weighted sequence with high B value and are markedly hypointense in the parametric ADC map (with values of 0.7 × 10−3 mm2/s). The cystic areas are hypointense on T1, hyperintense on T2, do not enhance and show facilitated water diffusion, with a large drop in signal with high B value and ADC of 2.2 × 10−3 mm2/s.")
, which show high signal on the ADC map due to the absence of restriction of water diffusion. These foci therefore lose the signal in the diffusion-weighted sequence with a high B value. In the dynamic study, with contrast, the periphery of the lesion shows intense enhancement, with the peak in the arterial phase of the study, while the centre, with cystic phenomena demonstrated by pathology examination, has little enhancement.")
. Centre row: diffusion sequence with B = 800 s/mm2 (enhanced in diffusion). Bottom row: apparent diffusion coefficient (ADC) map. A) 64-year-old female with hypertensive crises and paroxysms of headache. Right adrenal pheochromocytoma 22 mm in size with almost global cystic-haemorrhagic features, histologically demonstrated, with an antigravitational area with facilitated water diffusion and another gravitational (*) with low signal on T2 and T1 (not shown), relatively high in diffusion with B = 800 s/mm2 and low in the ADC map, with an average value of 0.9 × 10−3 mm2/s. The solid tissue wall, about 3 mm thick, shows high signal on T2 and diffusion restriction, with high signal at B = 800 and average ADC of 0.83. Ki-67 index in the surgical specimen: 2%. Has had no recurrence or progression 20 months later. B) A 49-year-old male with an incidental diagnosis of a left pheochromocytoma 43 mm in size confirmed by elevated serum and urine catecholamines and then histologically after laparoscopic resection. Predominantly solid mass, with two cystic areas (*) where water diffuses freely. In peripheral solid areas there is diffusion restriction, with an average ADC of 0.78 × 103 mm2/s. Ki-67 index: <1%. No recurrence or progression seven years later. C) 51-year-old female with hypertensive crises. Practically solid right pheochromocytoma 82 mm in size, with small anterior cystic foci (*) and facilitated diffusion of water. In solid areas there is a very high signal on T2 and moderate restriction of water diffusion, with an average ADC value of 0.94 × 10−3 mm2/s. Ki-67 index: 3%. No recurrence or progression six years later. D) 73-year-old male with grade II hypertension. Single left pheochromocytoma, practically solid, 51 mm in size, with very small central foci of cystic degeneration. Solid areas show high signal on T2 and relatively little restriction of water diffusion, with ADC of 1.3 × 10−3 mm2/s. Ki-67 index: 2%. No recurrence or progression a year after resection.")
 are shown in the top row and the pheochromocytoma (arrows) in the bottom row, both confirmed histologically. Two left-hand columns, CT: the number at the bottom right of each box corresponds to the mean attenuation value of the lesion in the axial slice shown. The adenoma has negative baseline attenuation, which allows it to be characterised. That of the pheochromocytoma is 30 HU, which is nonspecific. It enhances intensely in the arterial phase, heterogeneously (not shown), and in the portal phase, more homogeneously, exceeding 130 HU. No late washout study was performed. Remaining columns on the right, MRI: the adenoma shows a large drop in signal out-of-phase, which reveals its microscopic lipid content. The pheochromocytoma does not show a significant drop in signal and is surrounded by “India ink” artifact. The quantitative study also does not show a significant lipid content. The adenoma has an intermediate signal on T2. The pheochromocytoma shows a very high signal (\"light bulb sign\").")
CT technical protocol for the study of adrenal nodules.
| Collimation | Pitch Factor | Reconstruction thickness | Rotation time | Kilovolts | Milliamperes | Contrast | Vascular study phases |
|---|---|---|---|---|---|---|---|
| 0.6 mm | 0.6 | 1 mm | 0.5 s | 120 (100 or 80 in selected cases) | 158 baseline, automatic modulation | Iobitridol 350 mgI/mLRate: 3.5 cc/sAmount: 1.8 cc/kg or up to 140 ccBolus detection in abdominal aorta | Without contrastaLate arterial. Detection + 20 sPortal. Detection + 50 sDelayed. Detection +15 min |
Equipment: Somatom Definition AS+128®, Siemens Healthineers (Erlangen, Germany).
Basic sequences in our MRI study protocol for adrenal nodules (some parameters are adjusted to the patient's conditions).
| T1 in-phase/out-of-phase | T2 | Diffusion | T1 dynamic with contrast | |
|---|---|---|---|---|
| Sequence type | GE | TSE | EPI | VIBE |
| Orientation | Axial | Axial | Axial | Axial |
| Slice thickness (mm) | 3−5 | 2−5 | 5 | 3 |
| TR (ms) | 163 | 1600 | 1500 | 4.9 |
| TE (ms) | 2.4 / 4.8 | 75 | 79 | 2.39 |
| Reading FOV (mm) | 400 | 400 | 380 | 440 |
| Phase FOV (mm) | 75 | 75 | 75 | 72 |
| Technical details | Dual echo acquisition | Optional: fatsat or STIROptional: coronal | B 50, 400 and 800 mm2/sSPAIR fat suppressionADC map | Gadobutrol 1 mmol Gd/mL0.1 ml/kg at 2−2.5 ml/minPhases: 0 s-injection-1 min 06 s-1 min 49 s-2 min 26 s-4 min 12 sCan be left out if adenoma on dual echo T1 |
| Respiration | Apnoea | Navigation | Navigation | Successive apnoeas |
| Approx. time | 18−30 s | 3 min 29 s | 3 min 38 s | 5 × 11−18 s (up to 3 min 30 s) |
Magnetom Avanto® 1.5 T, Siemens Healthineers (Erlangen, Germany). The total examination time of the abbreviated study is around 10 min.
ADC: apparent diffusion coefficient; fatsat: spectral fat saturation; FOV: field of view; GE: gradient echo; EPI: echo-planar imaging; SPAIR: SPectral Attenuated Inversion Recovery; STIR: short inversion time inversion recovery; TE: echo time; TR: repetition time; TSE: turbo spin-echo; VIBE: volume interpolated breath-hold examination.
Generally it is impossible to predict histological aggressiveness.
The usefulness of radiomics in MRI scans to differentiate pheochromocytomas from other adrenal tumours has recently been analysed in several published papers28,29 (some multicentre.29) The application of radiomic analysis was studied by Kong et al29 using nomograms including clinical/analytical data, with promising results. However, a broader prospective evaluation of the results is necessary.
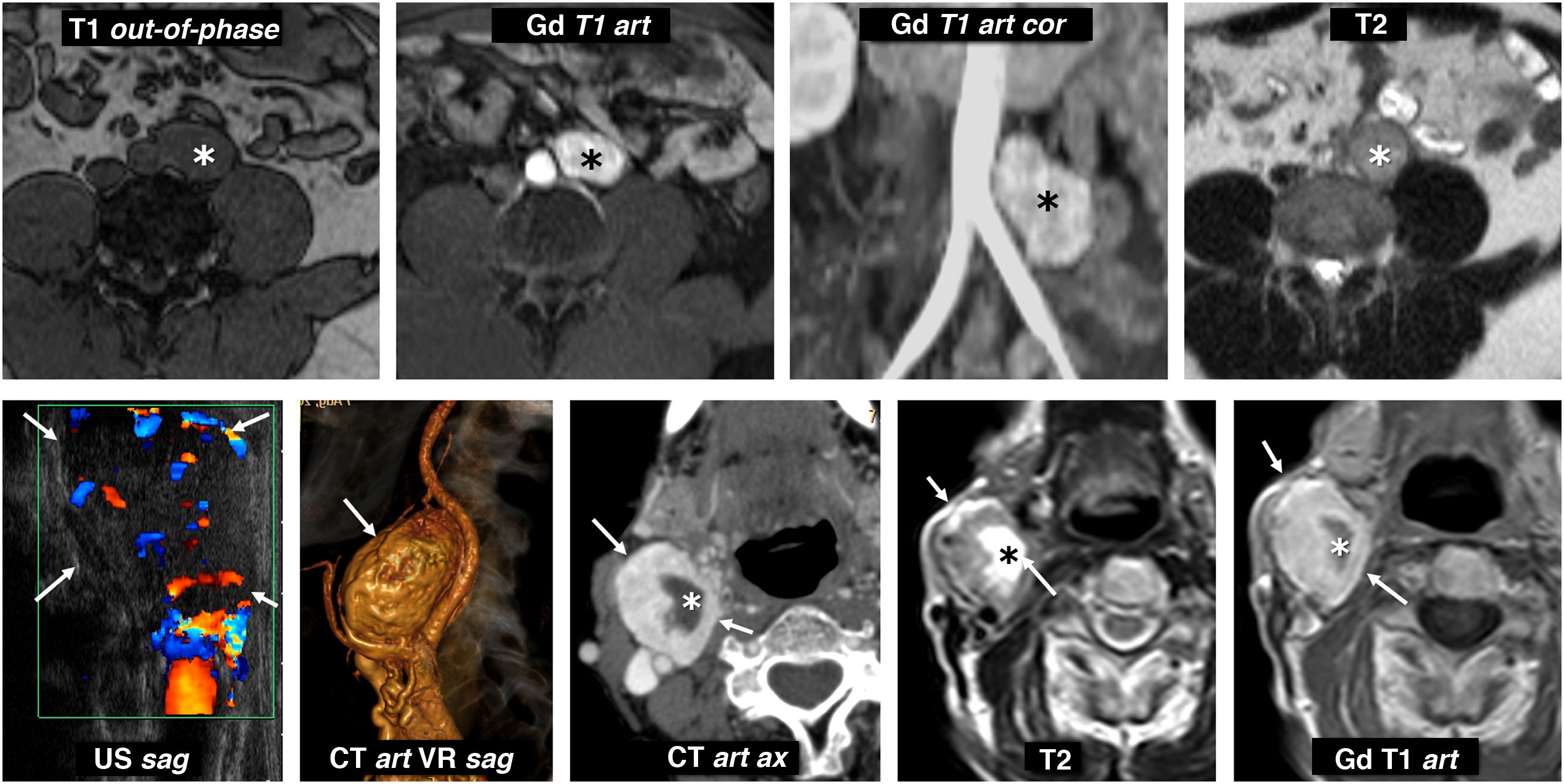
EAPGL show similar findings to those described, in different locations (Fig. 11).
 located to the left of the distal aorta, immediately below the origin of the inferior mesenteric artery, in the anatomical location of the organ of Zuckerkandl. This lesion is isointense on T1-weighted images and maintains its signal out-of-phase, is hyperintense on T2 and enhances very intensely in the arterial phase with Gadolinium. Bottom row: non-secretory parasympathetic paraganglioma of the carotid glomus. 46-year-old male. Consultation for palpable mass in neck. Intensely vascularised isoechoic nodular lesion (arrows) next to the right carotid bifurcation (the ultrasound image has been rotated 90° for better correlation). CT scan shows an intense enhancement, with some cystic foci inside (*). On MRI, solid areas show high signal on T2 and intermediate on T1, with intense and rapid enhancement. The cystic areas behave like water. Catecholamine determinations were normal.")
Extra-adrenal paragangliomas. Top row: metanephrine-producing sympathetic paraganglioma of the organ of Zuckerkandl. 31-year-old male investigated for hypertension and headache. Lobulated nodular lesion (*) located to the left of the distal aorta, immediately below the origin of the inferior mesenteric artery, in the anatomical location of the organ of Zuckerkandl. This lesion is isointense on T1-weighted images and maintains its signal out-of-phase, is hyperintense on T2 and enhances very intensely in the arterial phase with Gadolinium. Bottom row: non-secretory parasympathetic paraganglioma of the carotid glomus. 46-year-old male. Consultation for palpable mass in neck. Intensely vascularised isoechoic nodular lesion (arrows) next to the right carotid bifurcation (the ultrasound image has been rotated 90° for better correlation). CT scan shows an intense enhancement, with some cystic foci inside (*). On MRI, solid areas show high signal on T2 and intermediate on T1, with intense and rapid enhancement. The cystic areas behave like water. Catecholamine determinations were normal.
(Table 6)
Differential diagnosis by imaging of pheochromocytoma. Apart from these aspects, it is essential to consider the clinical and analytical context.
| Lesion | Possible confounding factors | Differential factors | |
|---|---|---|---|
| Malignant solid tumour | Metastasis | Possible cystic-necrotic phenomenaEnhancement overlap in hypervascular tumour metastasesNon-aggressive PCC consume glucoseNonspecific diffusion in general | Disseminated cancerMultiplicityLess and later enhancement in non-hypervascular tumoursLess signal intensity on T2-weighted images in solid areas |
| Carcinoma | Aggressive PCC can be invasive | Large, lobulated, heterogeneous, invasive massVery marked restriction of diffusion in solid areasLess and later enhancementLess signal intensity on T2-weighted images in solid areas | |
| Lymphoma | Frequent cystic-haemorrhagic phenomena | Generally lobulated and less well definedVery marked restriction of diffusion in solid areasLess and later enhancement | |
| Sarcoma | Frequent cystic-necrotic phenomena | Large, lobulated, heterogeneous, invasive mass | |
| Benign solid tumour | Adenoma | Possible cystic-haemorrhagic phenomena (uncommon)35% of PCC with similar late washout on CTCystic changes in PCC with baseline attenuation <10 HU | Signal drop out-of-phase in characteristic rangeLess and later enhancementLess signal intensity on T2-weighted images in solid areas |
| Myelolipoma | Well-defined round or oval lesions. Small percentage low in fat | Almost always macroscopic fat on CT or MRI in an areaExtremely rare for them to have cystic-necrotic phenomena | |
| Haemangioma | Well-defined round lesionsHigh signal on T2 | Characteristic enhancement pattern, similar to hepatic | |
| Benign cystic lesion | Cyst | Cystic areas by definition | No or almost no solid component |
| Haematoma-pseudocyst. | Cystic areas, sometimes thick-walled with calcifications | Previous medical historyVery little solid component, hypovascular or avascular | |
| Hydatid cyst | Variable presentation, with cystic areas if parasites viable | Frequent coexistence of hydatid disease of the liverDaughter vesicles or membranes are very characteristic | |
| Tuberculosis | Frequent cystic-necrotic areas, with peripheral enhancement | Bilateral involvementInvolvement of other organsNo solid tumour component |
PCC: pheochromocytoma.
(Fig. 12)
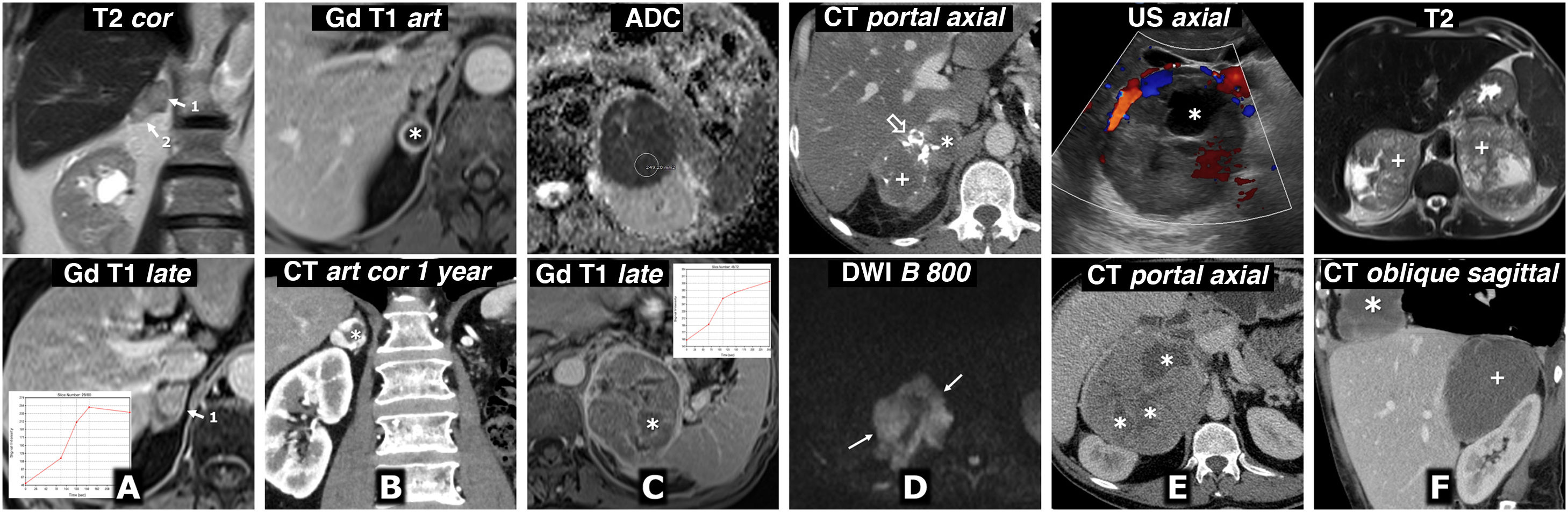
 Metastasis of squamous cell carcinoma of the lung. 72-year-old male. MRI: there are two solid right adrenal nodules (1 and 2), with moderately high signal on T2, marked restriction of water diffusion (not shown), without microscopic lipids (not shown), with relatively intense enhancement, although somewhat later than usual in pheochromocytoma. B) Metastasis of clear-cell renal cell carcinoma. 68-year-old male who had undergone surgery six years earlier for clear cell renal cell carcinoma in his left kidney. Almost completely cystic right adrenal nodule (*) with fine solid halo with intense enhancement in the arterial phase and free diffusion of water in its content (not shown). One year later, the lesion has grown and shows a greater solid component on CT, with very intense enhancement in arterial phase. Note the absence of the left kidney due to nephrectomy. The isolated radiological appearance of the lesion in both studies is indistinguishable from that of a pheochromocytoma. C) Primary adrenal carcinoma. 67-year-old male. MRI: large, well-defined, slightly lobulated left adrenal mass with a heterogeneous internal structure. The solid regions do not show a significant drop in their signal out-of-phase; they show hyperintensity in T2 (not shown), very marked restriction of water diffusion (ADC of 0.61 × 10−3 mm2/s) and slight (relatively less than 100%) and gradual enhancement. There are necrotic and haemorrhagic foci (*) confirmed histologically. D) Primary adrenal lymphoma. 51-year-old HIV-positive male. Lobulated and very heterogeneous right adrenal mass. It contains calcifications (hollow arrow), cystic (*) and haemorrhagic (+) areas, the latter with high signal on T1 and hypointense halo of haemosiderin on T2 (not shown). The solid areas show slight enhancement on CT and MRI (not shown) and intense restriction of water diffusion, with high signal with high B (arrows) and very low ADC values (not shown), around 0.68 × 10−3 mm2/s. The histological diagnosis was diffuse large B-cell lymphoma with activated immunophenotype (non-GCB) associated with the Epstein-Barr virus. E) Adrenal pleomorphic leiomyosarcoma. 40-year-old male. Large predominantly solid mass, although with cystic areas (*), lobulated and exerting significant compression on the inferior vena cava, which appeared invaded during surgery. F) Angiosarcoma with bilateral adrenal involvement (+) and a third mass in the right cardiophrenic angle (*). 53-year-old male. There are two adrenal masses and a supradiaphragmatic mass, well defined, with low density on CT, little enhancement, almost only peripheral and in some septa. On MRI they show cystic foci of various sizes, the odd small haemorrhagic focus and a significant restriction of water diffusion, with very low ADC values (not shown). The possibility of pheochromocytoma was raised in the context of a genetic syndrome, with a third lesion that could be metastatic or correspond to another tumour. The catecholamine study was normal. Core-needle biopsy was performed on one of the adrenal masses and the histological diagnosis was epithelioid angiosarcoma. It is impossible to determine which is the primary tumour.")
Differential diagnosis. Malignant adrenal lesions. A) Metastasis of squamous cell carcinoma of the lung. 72-year-old male. MRI: there are two solid right adrenal nodules (1 and 2), with moderately high signal on T2, marked restriction of water diffusion (not shown), without microscopic lipids (not shown), with relatively intense enhancement, although somewhat later than usual in pheochromocytoma. B) Metastasis of clear-cell renal cell carcinoma. 68-year-old male who had undergone surgery six years earlier for clear cell renal cell carcinoma in his left kidney. Almost completely cystic right adrenal nodule (*) with fine solid halo with intense enhancement in the arterial phase and free diffusion of water in its content (not shown). One year later, the lesion has grown and shows a greater solid component on CT, with very intense enhancement in arterial phase. Note the absence of the left kidney due to nephrectomy. The isolated radiological appearance of the lesion in both studies is indistinguishable from that of a pheochromocytoma. C) Primary adrenal carcinoma. 67-year-old male. MRI: large, well-defined, slightly lobulated left adrenal mass with a heterogeneous internal structure. The solid regions do not show a significant drop in their signal out-of-phase; they show hyperintensity in T2 (not shown), very marked restriction of water diffusion (ADC of 0.61 × 10−3 mm2/s) and slight (relatively less than 100%) and gradual enhancement. There are necrotic and haemorrhagic foci (*) confirmed histologically. D) Primary adrenal lymphoma. 51-year-old HIV-positive male. Lobulated and very heterogeneous right adrenal mass. It contains calcifications (hollow arrow), cystic (*) and haemorrhagic (+) areas, the latter with high signal on T1 and hypointense halo of haemosiderin on T2 (not shown). The solid areas show slight enhancement on CT and MRI (not shown) and intense restriction of water diffusion, with high signal with high B (arrows) and very low ADC values (not shown), around 0.68 × 10−3 mm2/s. The histological diagnosis was diffuse large B-cell lymphoma with activated immunophenotype (non-GCB) associated with the Epstein-Barr virus. E) Adrenal pleomorphic leiomyosarcoma. 40-year-old male. Large predominantly solid mass, although with cystic areas (*), lobulated and exerting significant compression on the inferior vena cava, which appeared invaded during surgery. F) Angiosarcoma with bilateral adrenal involvement (+) and a third mass in the right cardiophrenic angle (*). 53-year-old male. There are two adrenal masses and a supradiaphragmatic mass, well defined, with low density on CT, little enhancement, almost only peripheral and in some septa. On MRI they show cystic foci of various sizes, the odd small haemorrhagic focus and a significant restriction of water diffusion, with very low ADC values (not shown). The possibility of pheochromocytoma was raised in the context of a genetic syndrome, with a third lesion that could be metastatic or correspond to another tumour. The catecholamine study was normal. Core-needle biopsy was performed on one of the adrenal masses and the histological diagnosis was epithelioid angiosarcoma. It is impossible to determine which is the primary tumour.
Only 2–3% of incidentally found adrenal nodular lesions are malignant.
- -
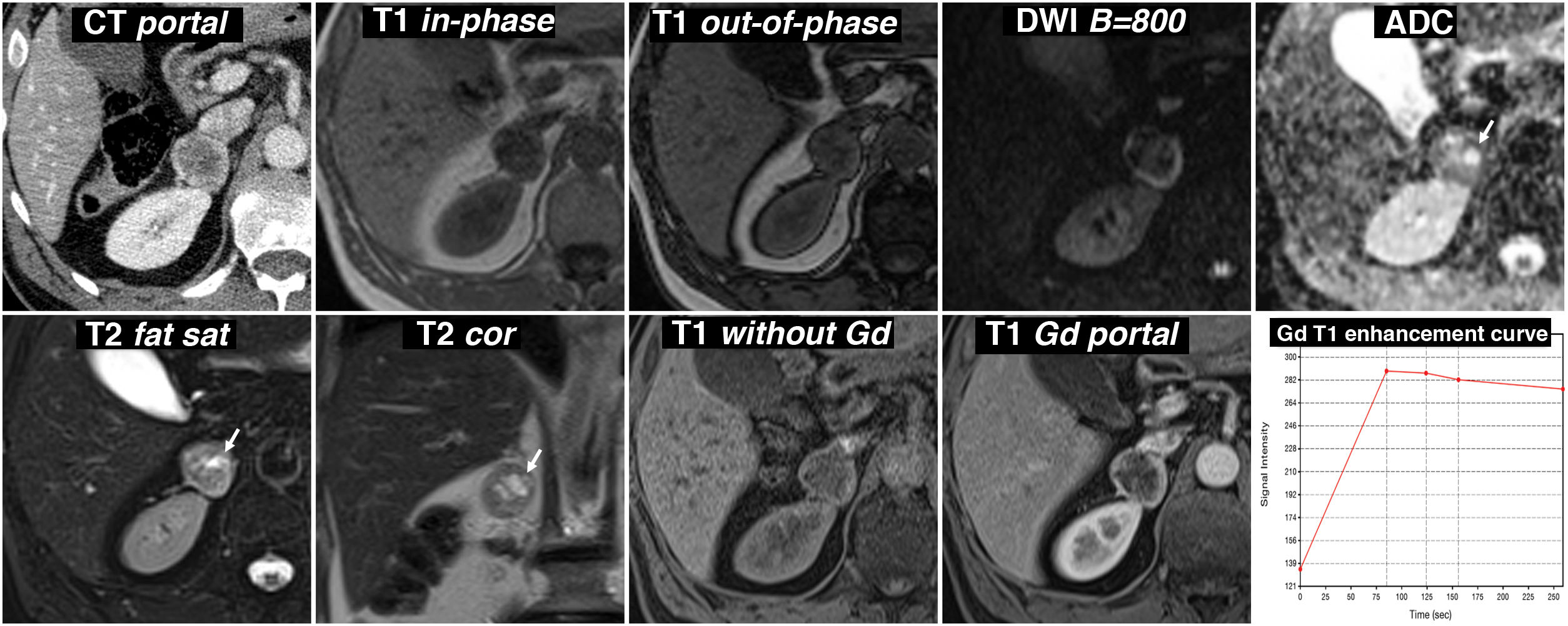
Metastasis. Various factors can make it difficult to differentiate between metastasis and pheochromocytoma in the context of neoplasia: limited value of the diffusion sequence; possible cystic-necrotic phenomena in metastases; overlapping enhancement pattern in cases such as neuroendocrine tumour or clear cell renal cell carcinoma; and marked metabolic activity of non-aggressive pheochromocytomas (Fig. 4). The multiplicity or the existence of disseminated disease can be useful in the differential diagnosis.
- -
Carcinoma. Uncommon, with a prevalence of 1–2 per million. In general, it behaves as a large, lobulated and heterogeneous mass, with necrotic, cystic and haemorrhagic areas, invasive behaviour, metastatic spread and later and less intense enhancement than pheochromocytoma. Solid areas show great restriction of water diffusion.
- -
Lymphoma. This is almost always secondary (in post-mortem examinations in 25% of lymphomas) and easy to diagnose in a context of typical extensive retroperitoneal involvement. Primary adrenal lymphoma is extremely rare and virtually impossible to diagnose with certainty from radiological imaging. It is usually heterogeneous, with cystic/haemorrhagic changes which can mimic pheochromocytoma, although with marked diffusion restriction and slight enhancement in solid areas.
- -
Sarcoma. Extremely rare. The origin of primary adrenal leiomyosarcomas is not fully understood. Like all other retroperitoneal leiomyosarcomas, it is thought they may originate in the muscle layer of venous structures (the adrenal vein or its tributaries). In general, they all behave like large, lobulated and heterogeneous adrenal masses, with cystic and necrotic areas, and some more invasive than others.
(Fig. 13)
- -
Adenoma. The drop in signal out-of-phase is characteristic up to a certain range, with this uncommon in pheochromocytomas. However, 35% of these show a similar late washout on CT (Fig. 6) and a small percentage of adenomas may have cystic or haemorrhagic changes which suggest the possibility of pheochromocytoma.
- -
Myelolipoma. Usually easy to diagnosis from the finding of macroscopic fat on CT or MRI. However, a small percentage of myelolipomas have very little fat component and it can be more difficult in these cases.
- -
Haemangioma. Rare (1/10,000 post-mortem examinations) and predominantly cavernous. They are small, well-defined lesions with behaviour similar to hepatic haemangiomas: high signal on T2-weighted sequence which can simulate pheochromocytoma, but peripheral globular enhancement with centripetal progression, occasionally incomplete. They may have phleboliths.
- -
Others. Oncocytoma, schwannoma, ganglioneuroma, adenomatoid tumour. Very rare. These may all contain cystic areas.
 Adenoma with cystic focal areas. 79-year-old female. CT in suspected constitutional syndrome: nodular lesion with cystic areas (*) in the left adrenal gland. On the MRI, which we cannot show due to the loss of the images in an unfortunate computer incident, a solid area with significant loss of signal out-of-phase was detected, which suggested adenoma with cystic changes. Seven years later, the lesion has not changed in two repeat CT scans. There are no clinical or laboratory data to suggest pheochromocytoma and no cancer has been diagnosed. B) Myelolipoma with a small amount of macroscopic fat. Incidental finding in a 42-year-old male being investigated for abdominal pain. The adrenal node (*) shows high signal on T2 and intense and rapid enhancement in most of the lesion, with little restriction of water diffusion (not shown). The diagnostic key is provided by the presence of an eccentric focus in the upper part (arrows), which loses signal in spectral fat saturation and STIR sequences; therefore, in relation to macroscopic fat. C) Adrenal haemangioma. 43-year-old female. Incidental finding of a well-defined right adrenal mass (arrows), with some rounded calcification on CT without contrast (hollow arrow). It shows little restriction of water diffusion, with relatively low signal intensity, although heterogeneous, with high B, starting from a very high signal on T2 (not shown). In the dynamic study with extracellular contrast, there is progressive centripetal filling-in following the vascular pool. The radiological suspicion of haemangioma was confirmed histologically.")
Differential diagnosis. Benign solid adrenal lesions. A) Adenoma with cystic focal areas. 79-year-old female. CT in suspected constitutional syndrome: nodular lesion with cystic areas (*) in the left adrenal gland. On the MRI, which we cannot show due to the loss of the images in an unfortunate computer incident, a solid area with significant loss of signal out-of-phase was detected, which suggested adenoma with cystic changes. Seven years later, the lesion has not changed in two repeat CT scans. There are no clinical or laboratory data to suggest pheochromocytoma and no cancer has been diagnosed. B) Myelolipoma with a small amount of macroscopic fat. Incidental finding in a 42-year-old male being investigated for abdominal pain. The adrenal node (*) shows high signal on T2 and intense and rapid enhancement in most of the lesion, with little restriction of water diffusion (not shown). The diagnostic key is provided by the presence of an eccentric focus in the upper part (arrows), which loses signal in spectral fat saturation and STIR sequences; therefore, in relation to macroscopic fat. C) Adrenal haemangioma. 43-year-old female. Incidental finding of a well-defined right adrenal mass (arrows), with some rounded calcification on CT without contrast (hollow arrow). It shows little restriction of water diffusion, with relatively low signal intensity, although heterogeneous, with high B, starting from a very high signal on T2 (not shown). In the dynamic study with extracellular contrast, there is progressive centripetal filling-in following the vascular pool. The radiological suspicion of haemangioma was confirmed histologically.
(Fig. 14)
- -
Adrenal cyst. Endothelial (some authors include lymphangiomas) or epithelial. They appear as well-defined thin-walled cystic masses, occasionally multiseptate.
- -
Haematoma-pseudocyst. Caused by blunt-force trauma (most often right), stress or severe illness (often bilateral). In the acute phase, it presents as a nodular lesion with high baseline attenuation and poorly defined peripheral areas. As it evolves, it usually shows a progressive reduction in attenuation and better delimitation until it forms a pseudocyst, usually with peripheral calcification.
- -
Hydatid cyst. Extremely rare, but the possibility should be considered in endemic areas. It may or may not coexist with liver disease or at other sites. It presents as a multicystic lesion. Diagnosis is easier when obvious daughter vesicles or membranes can be seen.
- -
Tuberculosis. Adrenal involvement is uncommon. It is usually bilateral, in the form of glandular thickening with central cystic or necrotic areas and peripheral enhancement. It can cause adrenal insufficiency and turn into residual calcifications.
 Haematoma and pseudocyst. Top image: 63-year-old male. Road-traffic accident. Traumatic left adrenal haemorrhage. Dense nodular lesion (58 HU) in the left adrenal gland (+), with dense blood-filled tracts towards the perirenal fat (arrows). In a later study, it had become a small pseudocyst (not shown). Bottom image: 42-year-old male. Adrenal pseudocyst. Incidental finding of a left adrenal cystic mass with peripheral calcification (*) in a patient investigated for a lymphoproliferative syndrome (see splenomegaly and a small left para-aortic adenopathy) with a remote history of trauma. B) Hydatid cyst. 56-year-old male with refractory hypertension and renal failure (study without contrast). Multicystic mass against the left adrenal gland. There was no hydatid disease in the liver or in other locations. Although cases of hydatid disease are still common in our health area, pheochromocytoma was initially suspected. Catecholamine studies were normal. The patient had surgery without incident, with hydatid cyst being diagnosed from the histology analysis. C) Tuberculosis. 57-year-old male, heavy drinker, recently diagnosed with pulmonary tuberculosis (not shown). A multicystic right adrenal mass can be seen (arrowheads), with a small extension to the inferior vena cava (hollow arrow). FNA of this lesion was performed, with microbiological confirmation of tuberculosis.")
Differential diagnosis. Benign cystic adrenal lesions. A) Haematoma and pseudocyst. Top image: 63-year-old male. Road-traffic accident. Traumatic left adrenal haemorrhage. Dense nodular lesion (58 HU) in the left adrenal gland (+), with dense blood-filled tracts towards the perirenal fat (arrows). In a later study, it had become a small pseudocyst (not shown). Bottom image: 42-year-old male. Adrenal pseudocyst. Incidental finding of a left adrenal cystic mass with peripheral calcification (*) in a patient investigated for a lymphoproliferative syndrome (see splenomegaly and a small left para-aortic adenopathy) with a remote history of trauma. B) Hydatid cyst. 56-year-old male with refractory hypertension and renal failure (study without contrast). Multicystic mass against the left adrenal gland. There was no hydatid disease in the liver or in other locations. Although cases of hydatid disease are still common in our health area, pheochromocytoma was initially suspected. Catecholamine studies were normal. The patient had surgery without incident, with hydatid cyst being diagnosed from the histology analysis. C) Tuberculosis. 57-year-old male, heavy drinker, recently diagnosed with pulmonary tuberculosis (not shown). A multicystic right adrenal mass can be seen (arrowheads), with a small extension to the inferior vena cava (hollow arrow). FNA of this lesion was performed, with microbiological confirmation of tuberculosis.
The indicated treatment is complete surgical resection, ideally laparoscopic, which needs to be performed with prior adrenergic blockade. The radiologist should take part in the planning,3,5–8,12,14,24,30 jointly reviewing the vascular anatomy (particularly venous) and providing helpful reconstructions (Fig. 15). In cases of hereditary syndrome, conservative resection of the gland should be attempted. As genetic and molecular characterisation advances, local or systemic radiometabolic therapies are being developed to be applied on an individual basis.7,8,12,14
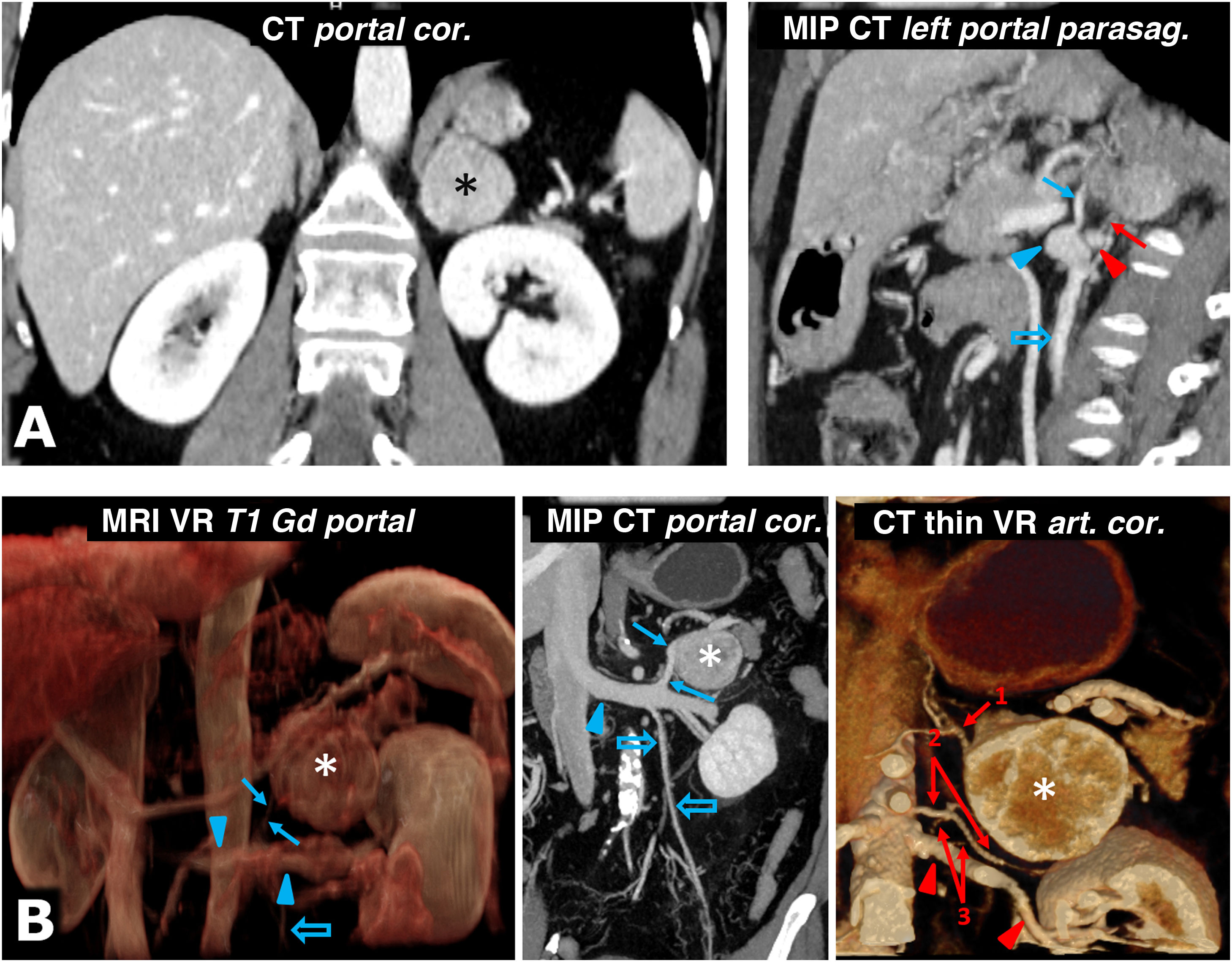
 57-year-old female. Incidental finding on ultrasound (not shown) for haematuria of well-defined solid left adrenal mass (*), 38 mm in size, in the left adrenal gland, with intense enhancement and late lavage similar to the adenoma (not shown). The results of the catecholamine tests were abnormal, and it was decided to intervene. Shortly before laparoscopic surgery, the vascular anatomy was reviewed with the surgeons, locating the superior suprarenal arteries (almost always branch of the inferior phrenic), middle suprarenal arteries (normally a direct branch of the aorta) and inferior suprarenal arteries (red arrow; generally a branch of the ipsilateral renal artery), as well as the suprarenal vein (blue arrow; usually single and tributary of the renal vein on the left side and the inferior vena cava on the right) and all pertinent anatomical details. Red arrowhead: left renal artery. Blue arrowhead: left renal vein. Blue hollow arrow: left ovarian vein. Laparoscopic resection was performed without incident. The tumour met histological criteria for aggressiveness (PASS score of 6). B) 73-year-old male with grade II hypertension. The MRI volumetric reconstruction shows the relationships between the left adrenal pheochromocytoma (*), the adrenal vein (blue arrow), and the left renal vein into which it empties (blue arrowheads). Blue hollow arrow: spermatic vein. CT reconstructions provide more precise anatomical details for surgical planning. Red arrows: left adrenal arteries: 1: superior; 2: middle, and 3: lower (2 arteries as anatomical variant). Red arrowheads: left renal artery.")
Help in surgical planning. A) 57-year-old female. Incidental finding on ultrasound (not shown) for haematuria of well-defined solid left adrenal mass (*), 38 mm in size, in the left adrenal gland, with intense enhancement and late lavage similar to the adenoma (not shown). The results of the catecholamine tests were abnormal, and it was decided to intervene. Shortly before laparoscopic surgery, the vascular anatomy was reviewed with the surgeons, locating the superior suprarenal arteries (almost always branch of the inferior phrenic), middle suprarenal arteries (normally a direct branch of the aorta) and inferior suprarenal arteries (red arrow; generally a branch of the ipsilateral renal artery), as well as the suprarenal vein (blue arrow; usually single and tributary of the renal vein on the left side and the inferior vena cava on the right) and all pertinent anatomical details. Red arrowhead: left renal artery. Blue arrowhead: left renal vein. Blue hollow arrow: left ovarian vein. Laparoscopic resection was performed without incident. The tumour met histological criteria for aggressiveness (PASS score of 6). B) 73-year-old male with grade II hypertension. The MRI volumetric reconstruction shows the relationships between the left adrenal pheochromocytoma (*), the adrenal vein (blue arrow), and the left renal vein into which it empties (blue arrowheads). Blue hollow arrow: spermatic vein. CT reconstructions provide more precise anatomical details for surgical planning. Red arrows: left adrenal arteries: 1: superior; 2: middle, and 3: lower (2 arteries as anatomical variant). Red arrowheads: left renal artery.
Local recurrence after surgical resection is possible, even many years later. Long-term follow-up is recommended, for at least ten years after the operation and in certain situations for life.3,6–8
ConclusionsAlthough the clinical presentation and laboratory abnormalities can guide the diagnosis of pheochromocytoma, it is not uncommon for the radiologist to be the first to suggest it from the findings in an imaging test.
We need to remember that the characteristic presentation is a well-defined adrenal nodular lesion which has a high signal intensity on T2-weighted images, rapid and intense enhancement and, frequently, cystic and haemorrhagic features.
In uncertain cases, measurement of catecholamines or additional Nuclear Medicine tests may be recommended.
Percutaneous puncture should be avoided due to possible potentially serious secondary complications.
Pheochromocytomas can occur in a number of different hereditary syndromes, notably von Hippel-Lindau, MEN 2 and hereditary paraganglioma. In sporadic cases, there are also very often underlying genetic abnormalities, which are changing the paradigm of the disease.
It is important to have a good understanding of the numerous lesions with which the differential diagnosis can be made.
Authorship- 1
Person responsible for the integrity of the study: MACC.
- 2
Study concept: MACC.
- 3
Study design: MACC.
- 4
Data collection: MACC, JEI, GCFP, MRC and AF.
- 5
Data analysis and interpretation: MACC, JEI, GCFP, MRC and AF.
- 6
Statistical processing: not applicable.
- 7
Literature search: MACC and JEI.
- 8
Drafting of the article: MACC, JEI and GCFP.
- 9
Critical review of the manuscript with relevant intellectual contributions: MACC, JEI, GCFP, MRC and AF.
- 10
Approval of the final version: MACC, JEI, GCFP, MRC and AF.
The authors declare that they have no conflicts of interest.
Please cite this article as: Corral de la Calle MA, Encinas de la Iglesia J, Fernández-Pérez GC, Repollés Cobaleda M, Fraino A. Feocromocitoma adrenal. Claves para el diagnóstico radiológico. Radiología. 2022;64:348–367.

