




Connective tissue diseases are often associated with lung diseases that lead to high morbidity and mortality, including interstitial disease, airway disease, pleural lesions, and vascular disease. High resolution CT offers high sensitivity for detecting parenchymal disease and potentially reversible lesions, thus helping to guide treatment. This article emphasizes interstitial pneumonia in association with connective tissue disease and the characteristics that differentiate this entity from idiopathic types. Likewise, we review the most common pulmonary manifestations of each connective tissue disease with the aim of providing the radiologist with a practical approach to the diagnosis and management of these diseases in daily clinical practice.
Las enfermedades del tejido conectivo se asocian con frecuencia a un amplio número de patologías como las enfermedades intersticiales y de la vía aérea, las lesiones pleurales y la patología vascular, que presentan una alta morbilidad y mortalidad en estos pacientes. La tomografía computarizada de alta resolución tiene una alta sensibilidad en la detección de la enfermedad parenquimatosa y de la vía aérea, lo que permite diagnosticar la fibrosis establecida, o bien lesiones potencialmente reversibles, y facilita la correcta indicación del tratamiento en estos pacientes. En este artículo se hace una especial incidencia en las neumonías intersticiales asociadas a las enfermedades del tejido conectivo y en sus características diferenciales con respecto a las formas idiopáticas. También, se revisan las manifestaciones pulmonares más frecuentes en cada conectivopatía, con la intención de proporcionar al radiólogo un enfoque práctico en el diagnóstico y manejo de estas afecciones en la clínica diaria.
Connective tissue diseases (CTD) are often associated with a wide range of lung conditions that result in significant morbidity and mortality. These associations are varied, ranging from parenchymal, pleural and airway lung disease, to vascular, heart and musculoskeletal involvement; although each CTD is usually associated with a specific entity.1 For this reason, the presence of lesions related to the underlying CTD such as joint erosions, esophageal dilation, or enlargement of the pulmonary artery may assist the radiologist in the diagnosis of lung lesions. However, in some of the patients, the lung disease may precede the clinical symptoms of the collagen disease.1 In addition, since these patients are immunocompromised, infections are one of the most common causes of respiratory disease, and they should be considered in the evaluation of lung abnormalities on chest imaging in patients with ETC.2 Lastly, drug-related adverse reactions should also be included in the differential diagnosis.
Therefore, the diagnosis and clinical management of these patients is complex and should be based on the combination of different diagnostic approaches: clinical symptoms, laboratory and imaging findings.2
The aim of this paper is to review the pulmonary manifestations associated with the CTDs, with an emphasis on interstitial and airway disease, and on the high-resolution computed tomography (HRCT) findings, which facilitate early diagnosis of these conditions.
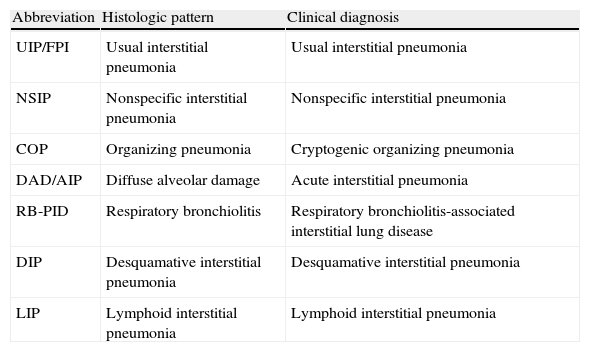
Interstitial lung disease associated with connective tissue diseasesDiffuse interstitial lung diseases are a common manifestation of CTDs, with histologic and radiologic patterns similar to those found in the idiopathic forms. Therefore, diffuse interstitial lung diseases can be classified using the Consensus Classification of the Idiopathic Interstitial Pneumonias (IIP) of the ATS/ERS 20023 (Table 1). The pneumonias most commonly associated with CTDs are the nonspecific interstitial pneumonia (NSIP) and the usual interstitial pneumonia (UIP), common in scleroderma (SCL) and rheumatoid arthritis (RA), respectively.4,5 Organizing pneumonia is also commonly found in CTDs, usually in association with other histologic patterns of interstitial pneumonia.6 Lymphoid interstitial pneumonia (LIP) is found in Sjögren's syndrome (SjS).7 The pattern of diffuse alveolar damage (DAD)3,8 is seen in acute exacerbations of pulmonary fibrosis and lupus-related pneumonia. Desquamative interstitial pneumonia (DIP) does not have a clear correlation with CTDs, but a few cases of DIP associated with RA have been reported.1
Current classification of Idiopathic Interstitial Pneumonias. Correlation between histologic pattern and clinical diagnosis.
| Abbreviation | Histologic pattern | Clinical diagnosis |
| UIP/FPI | Usual interstitial pneumonia | Usual interstitial pneumonia |
| NSIP | Nonspecific interstitial pneumonia | Nonspecific interstitial pneumonia |
| COP | Organizing pneumonia | Cryptogenic organizing pneumonia |
| DAD/AIP | Diffuse alveolar damage | Acute interstitial pneumonia |
| RB-PID | Respiratory bronchiolitis | Respiratory bronchiolitis-associated interstitial lung disease |
| DIP | Desquamative interstitial pneumonia | Desquamative interstitial pneumonia |
| LIP | Lymphoid interstitial pneumonia | Lymphoid interstitial pneumonia |
Each entity is defined by a characteristic histologic pattern; however, except for pulmonary fibrosis with UIP pattern on HRCT, the histologic pattern is not correlated with a sufficiently defined radiologic pattern that would allow the radiologist to make a confident diagnosis.3,9 Because of the overlap of HRCT features between the different entities, surgical biopsy is needed to make a confident diganosis.3
The frequency of appearance of IIPs in the different CTDs is summarized in Table 2. The most common radiologic presentation of each IIP is shown in Table 3.
Frequency of parenchymal involvement in the different connective tissue diseases.
| RA | SLE | SCL | DM/PM | SjS | MCTD | |
| UIP | ++ | + | + | ++ | + | + |
| NSIP | + | + | +++ | +++ | ++ | +++ |
| COP | + | + | ++ | + | ||
| LIP | ++ | + | ||||
| DAD | + | ++ | + | ++ | ||
| Bronchiolitis | +++ | ++ |
RA: rheumatoid arthritis; DAD: diffuse alveolar damage; DM/PM: dermato/polymyositis; MCTD: mixed connective tissue disease; SCL: scleroderma: SLE: systemic lupus erythematosus; LIP: lymphoid interstitial pneumonia; NSIP: nonspecific interstitial pneumonia; UIP: usual interstitial pneumonia; COP: cryptogenic organizing pneumonia; SSJ: Sjögren's syndrome.
+: less common; ++: common; +++: more common.
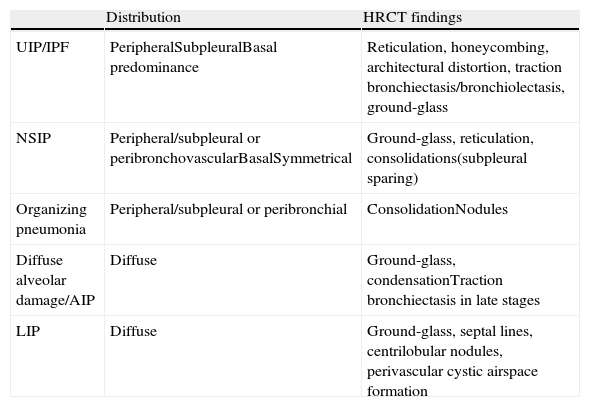
Radiological presentation of interstitial pneumonias associated with connective tissue diseases.
| Distribution | HRCT findings | |
| UIP/IPF | PeripheralSubpleuralBasal predominance | Reticulation, honeycombing, architectural distortion, traction bronchiectasis/bronchiolectasis, ground-glass |
| NSIP | Peripheral/subpleural or peribronchovascularBasalSymmetrical | Ground-glass, reticulation, consolidations(subpleural sparing) |
| Organizing pneumonia | Peripheral/subpleural or peribronchial | ConsolidationNodules |
| Diffuse alveolar damage/AIP | Diffuse | Ground-glass, condensationTraction bronchiectasis in late stages |
| LIP | Diffuse | Ground-glass, septal lines, centrilobular nodules, perivascular cystic airspace formation |
IPF: idiopathic pulmonary fibrosis; AIP: acute interstitial pneumonia; LIP: lymphoid interstitial pneumonia; NSIP: nonspecific interstitial pneumonia; UIP: usual interstitial pneumonia.
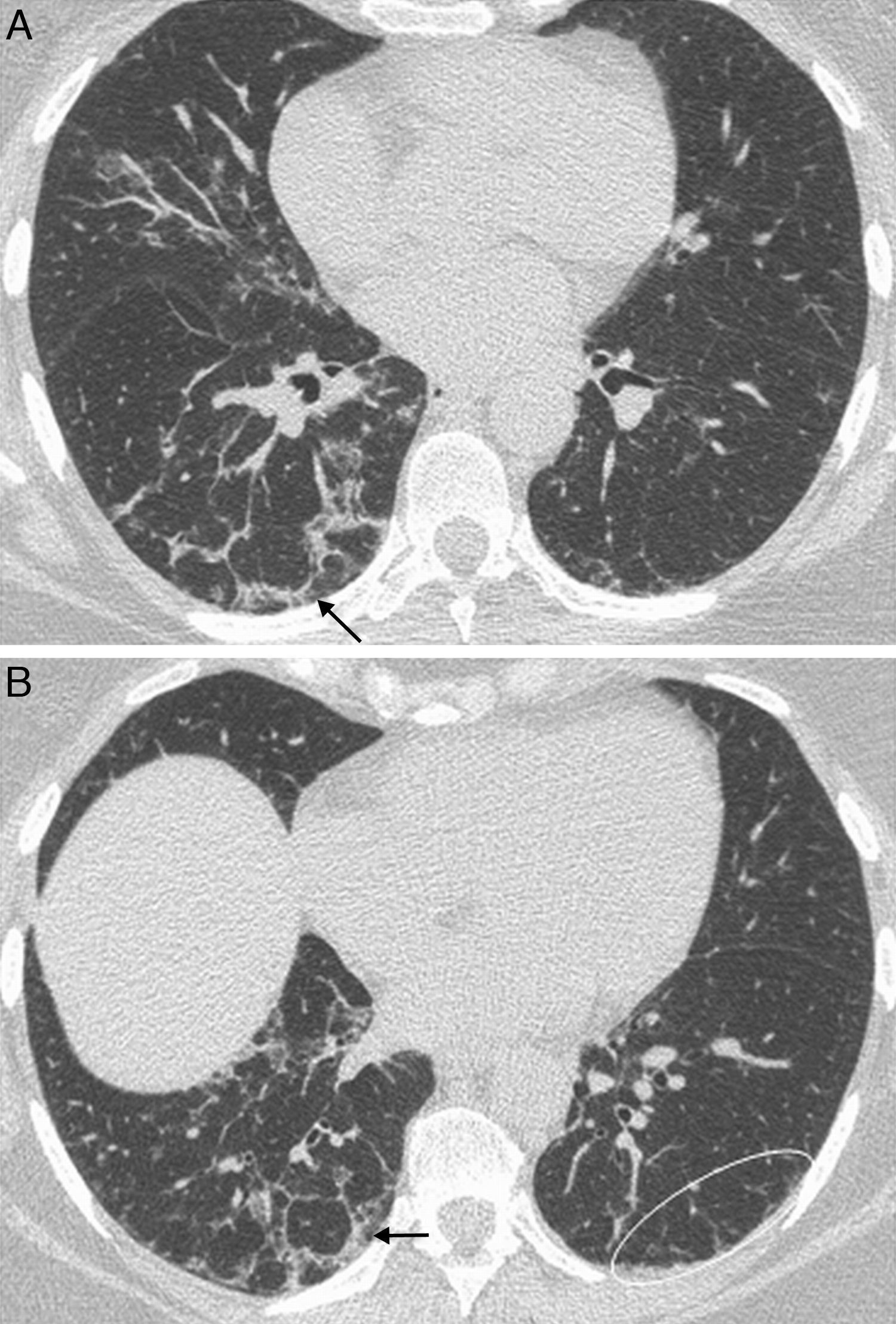
Histologic features of UIP include a heterogeneous, patchy pattern where areas of normal parenchyma alternate with areas at different stages with inflammation, fibroblastic foci, fibrosis and honeycombing.3 This “temporal heterogeneity” is characteristic of the UIP and it helps distinguish it from NSIP.3 Lesions are predominantly basal and subpleural, and in most cases there is honeycombing at diagnosis; therefore, absence of honeycombing makes UIP unlikely and should lead to an alternative diagnosis.10 The typical radiologic features, which correlate with the histologic appearance, include bilateral and symmetrical reticulation, with subpleural and posterior basal predominance.11 Honeycombing is a common finding that supports the diagnosis of UIP,12,13 while ground-glass attenuation is rare or nonexistent (Fig. 1). UIP is the histologic pattern most commonly seen in patients with rheumatoid arthritis.2,5,14
 and honeycombing (arrows).")
Interstitial lung disease with radiologic pattern of UIP in a male patient with long-standing rheumatoid arthritis. Axial HRCT image shows bilateral and symmetrical reticulation, with subpleural and basal predominance, associated with traction bronchiectasis (arrowheads) and honeycombing (arrows).
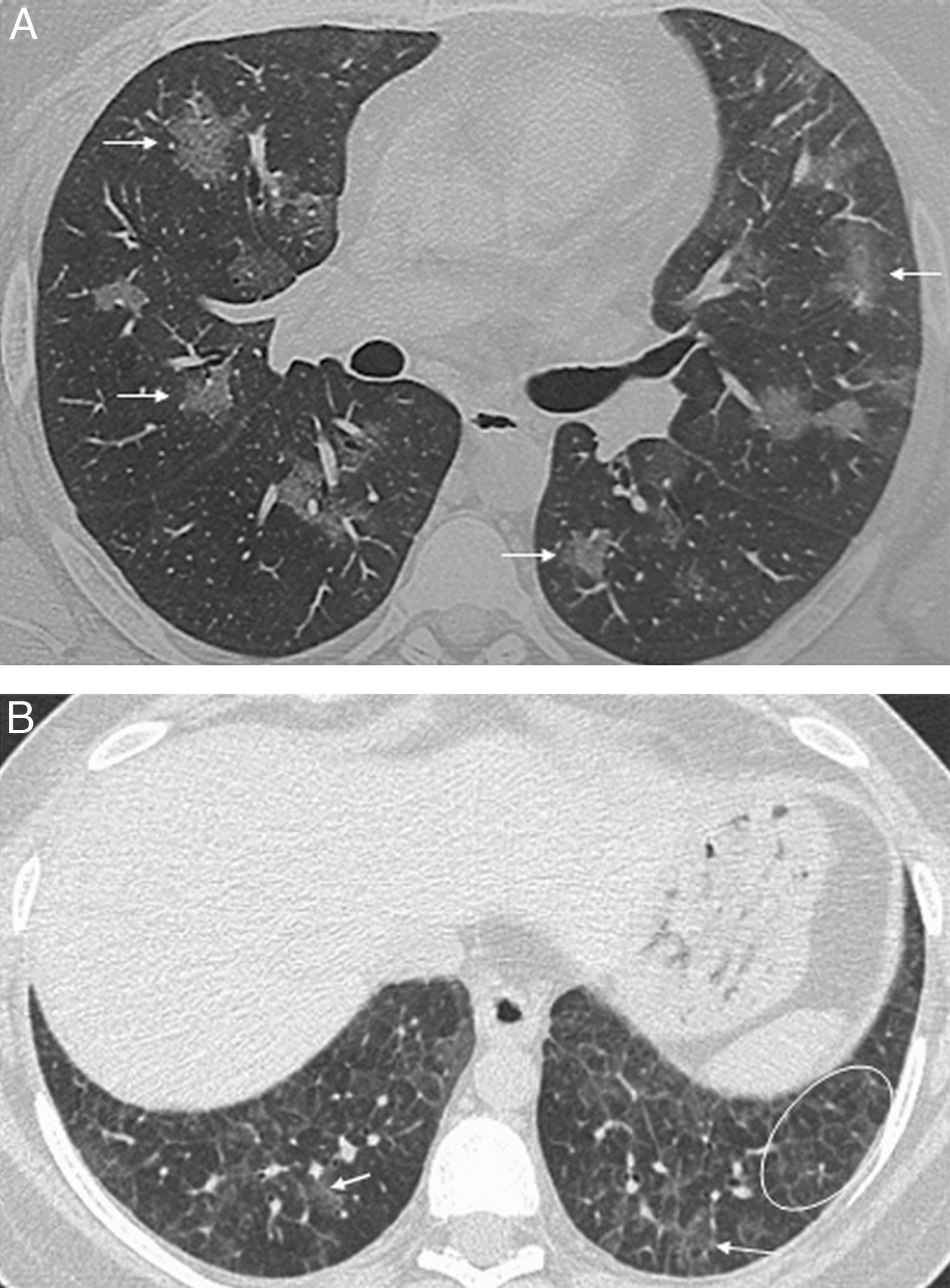
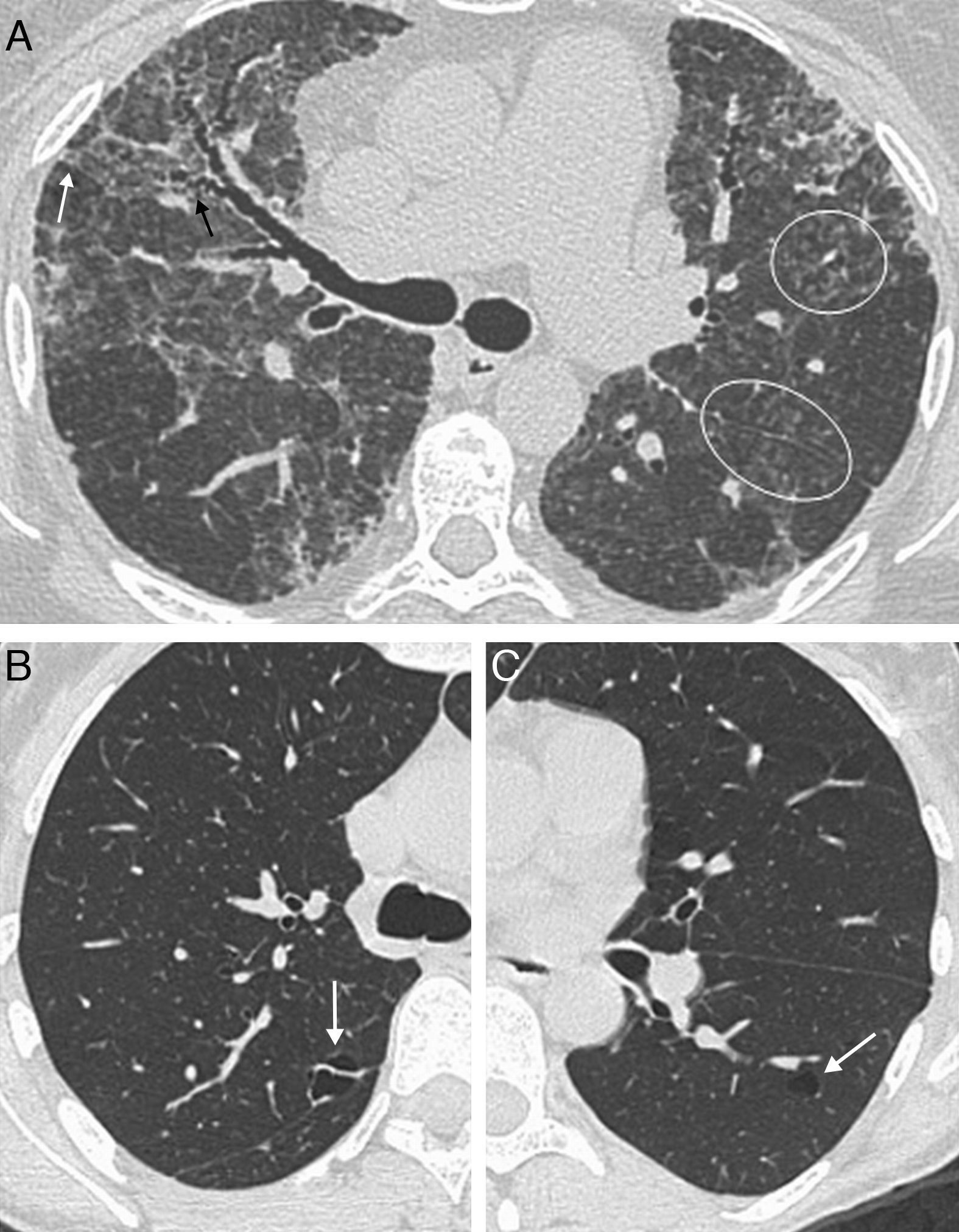
Unlike UIP, the typical histologic feature of NSIP is temporal and spatial homogeneity. Changes range from predominant inflammation, with uniform thickening of the alveolar septa and infiltrate of lymphocytes and plasma cells (cellular NSIP), to uniform interstitial thickening with preservation of the alveolar architecture (fibrotic NSIP).8,13,15 The radiologic pattern may overlap with that of UIP, but ground-glass attenuation is usually more common in NSIP and is the salient feature of cellular NSIP,11,13 and honeycombing is rare at diagnosis.7,12 Lower lobe predominance with peribronchovascular distribution and subpleural sparing is very suggestive of NSIP7,13 (Fig. 2). This is the most common interstitial pneumonia associated with CTDs,6 and it is been suggested that NSIP is a lung manifestation of undifferentiated connective tissue disease.16 While this may be the case in some patients, to date, no clear evidence has validated this hypothesis, and the subject remains open to debate, pending further research.15,17
, associated with traction bronchiectasis (arrowheads). Subpleural sparing is suggestive of NSIP (long arrows).")
The term “lung-dominant CTS” or “autoimmune-featured interstitial lung disease” has been recently proposed, encompassing those patients with interstitial disease, positive antibodies and histologic features of CTD, but do not meet the diagnostic criteria for a specific CTD.18 These patients usually show a histologic and radiologic pattern of UIP, and have a prognosis similar to those with idiopathic pulmonary fibrosis and worse than those with interstitial pneumonia associated with a CTD.19
One characteristic of the interstitial pneumonias in patients with CTD is the presence of coexisting histologic patterns on the same biopsy specimen,6 a finding that is, according to some authors, very suggestive of CTD.6,20
The prognosis of the interstitial pneumonias associated with CTD is usually better than that of the idiopathic forms. This would explain, in part, why UIP is the most common histologic pattern in IIPs, while the most common pattern in CTD-associated pneumonias is NSIP,4,6,21 which has a more favorable prognosis. In addition, the prognosis of the UIP pattern associated with a CTD is better than UIP with idiopathic pulmonary fibrosis.4,21,22 The histologic pattern differs between the two groups, with less extensive honeycombing and fibroblastic foci, and a higher rate of germinal and inflammatory centers in patients with a CTD than with idiopathic pulmonary fibrosis.22,23 However, no significant differences have been found between the prognosis of patients with NSIP associated with a CTD and those with idiopathic NSIP.21 Clinical findings and prognosis of organizing pneumonia associated with a CTD are similar to those of idiopathic organizing pneumonia; however, the former tends to recur and HDCT usually shows traction bronchiectasis.24
Acute exacerbation of interstitial pneumonia, similar to that of idiopathic pulmonary fibrosis, also occurs in interstitial pneumonias associated with a CTD and has been reported in patients with dermatomyositis (DM), RA, SCL and systemic lupus erythematosus (SLE). Acute exacerbation is more common and has poorer prognosis in RA with histologic pattern of UIP and in DM.25,26 The histologic pattern is that of DAD, associated or not with organizing pneumonia.8 HRCT shows ground-glass attenuation and de novo peripheral, multifocal or diffuse consolidation.8 Given its high mortality rates, this condition should be considered by the radiologist in the presence of suggestive radiologic changes in patients with CTD and interstitial lung disease.25
Sensitivity of chest radiography for the detection of interstitial lung disease is very low. In addition, the clinical symptoms are not useful in the evaluation of interstitial disease since dyspnea may be caused by a number of factors and lung function tests are not representative in the early stages of the disease.2 Conversely, HRCT provides good sensitivity and specificity and is able to provide a confident diagnosis of interstitial lung disease and fibrosis.2 In addition, this technique allows multiplanar reformatted images and has the advantage of being an affordable and non-invasive technique. The main limitation is the lack of reliable criteria for establishing a correlation between the extent of the lesions and the presence of subclinical or clinically significant disease.2,27 HRCT also plays in important role in the detection of signs suggestive of potentially reversible disease. As in IIPs, the presence of ground-glass opacities without signs of fibrosis (traction bronchiectasis and honeycombing) is suggestive of reversibility and good response to treatment.28,29 On the other hand, the absence of ground-glass opacity and presence of signs of fibrosis on HRCT should avoid unsuccessful treatments with potentially toxic drugs.2
A multidisciplinary approach to lung disease associated with CTD is paramount for an accurate diagnosis and management of these patients. The high diagnostic accuracy of HRCT, in conjunction with an adequate evaluation of the clinical and functional features, bronchoalveolar lavage (BAL) and laboratory studies make that lung biopsy could be primarily reserved for cases with atypical clinical or radiologic presentation.30
Other lung conditions associated with connective tissue diseasesInfectionsIt should be highlighted that lung infections are common in patients with CTD due to their impaired immunity. For this reason, infections should be ruled out when lung consolidation or nodules are detected. The most common infections are bacterial, and are a major cause of death in patients with SLE.31 Immunosuppressive drugs and the new generation of biologic agents used to treat rheumatic diseases have resulted in an increased incidence of infections caused by opportunistic pathogens, especially Mycobacterium tuberculosis and atypical mycobacteria,32 fungi and Pneumocistis jiroveci.1,2 Screening chest radiographs and BAL are indicated and fungal or mycobacterial infection should be suspected in the presence of parenchymal abnormalities in these patients.1
Adverse drug reactionsMany of the drugs used to treat CTDs result in pulmonary side effects, rendering it more difficult to evaluate radiologic findings in these patients. This occurs more frequently in RA. Pulmonary toxicity caused by methotrexate manifests within months of starting therapy in 5–10% of patients, with NSIP being the most common pattern.33
The new biologic agents (monoclonal antibodies), especially anti-TNF, have been related to diffuse interstitial lung disease (mainly UIP and NSIP), sarcoid-like lesions, acute exacerbation of interstitial disease, pulmonary vasculitis, hypersensitivity pneumonitis and pneumonias with negative cultures, among others.34,35
Lung neoplasmsPatients with CTDs have an increased risk of lung cancer, favored by pulmonary fibrosis, common in RA and SCL, or by the paraneoplastic etiology of DM/PM.2,36 A higher incidence of lymphoma has also been identified in patients with RA and SSj.37 Therefore, histologic diagnosis is required to rule out malignancy when new lung nodules are identified in these patients.37
Radiologic findings in lung disease associated with different connective tissue diseasesRheumatoid arthritisRA is often related to lung abnormalities on HRCT, although respiratory symptoms occur later in the course of the disease.
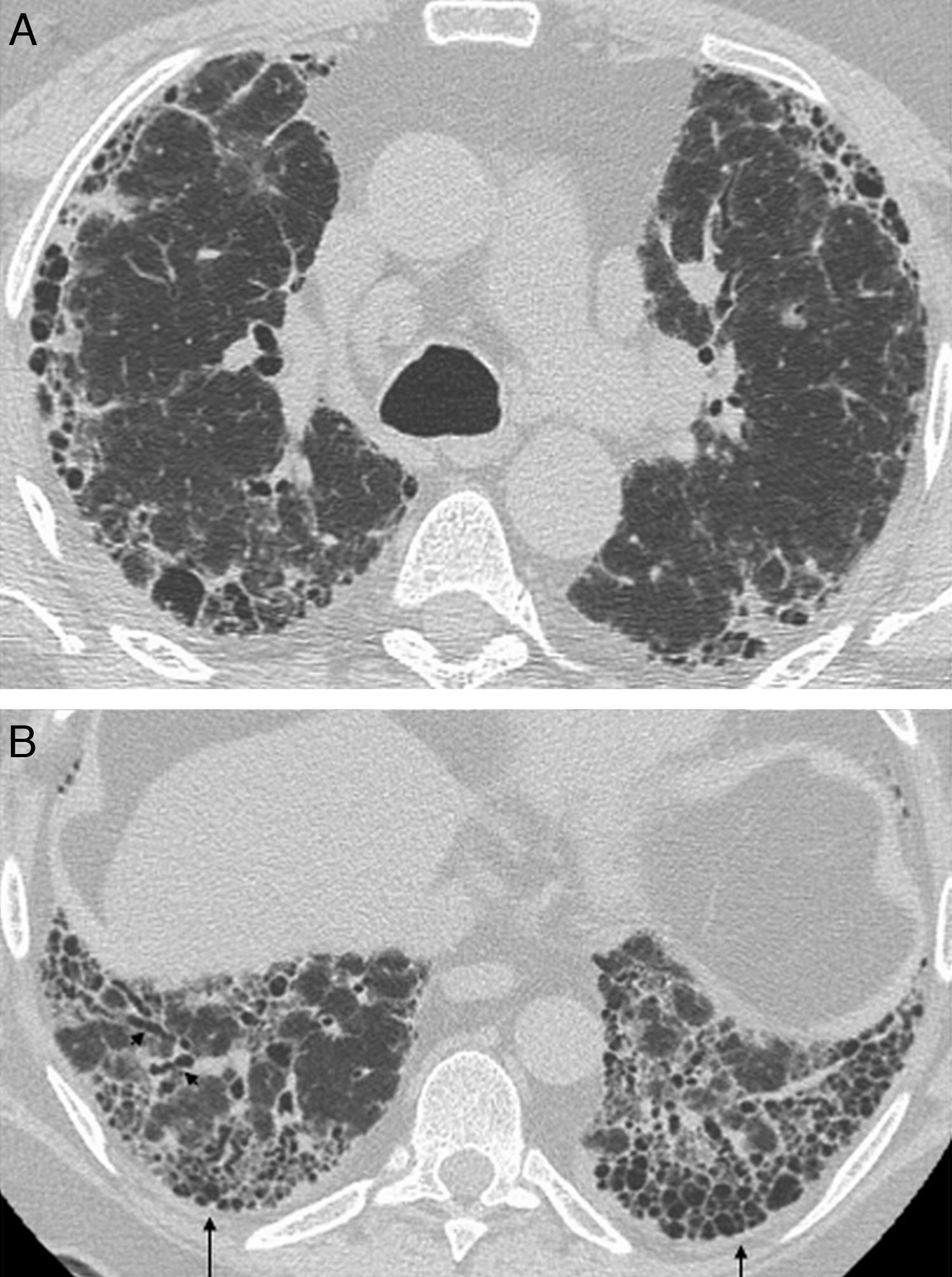
The earliest pulmonary manifestation is airway disease, being constrictive bronchiolitis and follicular bronchiolitis the most common histologic patterns.6,38 Cylindrical bronchiectasis is also a common finding in patients with RA, associated or not with bronchiolitis.5,37 Constrictive bronchiolitis is characterized by concentric peribronchiolar fibrosis with luminal narrowing and obliteration.39 In RA, constrictive bronchiolitis may appear associated or not with use of penicillamine therapy.40 Follicular bronchiolitis is a type of cellular bronchiolitis characterized by the presence of lymphoid follicles with bronchial and peribronchial distribution41 and it is seen in patients with CTDs, particularly AR6 and SSj.40 HRCT shows mosaic perfusion and air trapping,1,42 associated with cylindrical bronchiectasis and bronchiolectasis (Fig. 3). Centrilobular nodules are also seen in follicular bronchiolitis, an evidence of bronchiolar and peribronchiolar lesion.40,42 Expiratory HRCT is always recommended in these patients to rule out air trapping, indicative of bronchiolitis, which may be subclinical in many patients1 (Fig. 4).
 and centrilobular nodules with a patchy distribution (black arrows).")
 Inspiratory HRCT image shows no abnormalities. (B) Expiratory HRCT shows mosaic pattern with bilateral radiolucent areas corresponding to air trapping and consistent with bronchiolitis (arrows).")
Male patient with rheumatoid arthritis who presents with dyspnea. Inspiratory and expiratory axial HRCT. (A) Inspiratory HRCT image shows no abnormalities. (B) Expiratory HRCT shows mosaic pattern with bilateral radiolucent areas corresponding to air trapping and consistent with bronchiolitis (arrows).
Interstitial lung disease is more common in men1 and usually appears once the clinical symptoms of RA are established, but in some of the patients, the lung disease precedes RA.14 It is usually associated with restriction of respiratory function parameters.14 Several studies have confirmed that the radiologic pattern of UIP is more common than NSIP in patients with AR,43 unlike the rest of CTDs. The most common HRCT finding is the presence of peripheral reticulation associated with varying degrees of honeycombing, depending on the series2 (Fig. 1). In a series of 63 patients with pulmonary disease associated with RA, Tanaka et al.5 reported reticulation on HRCT in 98% of cases and ground-glass opacity in 90%. The radiologic pattern of UIP was the most common (26 patients), followed by NSIP (19 patients), bronchiolitis (11 patients) and organizing pneumonia (5 patients). In 13 of the 17 patients with available histologic analysis, the histologic pattern correlated with the radiologic findings. In another series with 18 RA patients who underwent surgical biopsy for suspected interstitial lung disease,14 UIP was the most common histologic pattern, with a higher incidence in men and smokers, while NSIP was seen more frequently in women and non-smokers. RA patients with UIP pattern have worse prognosis than those with NSIP.43,44
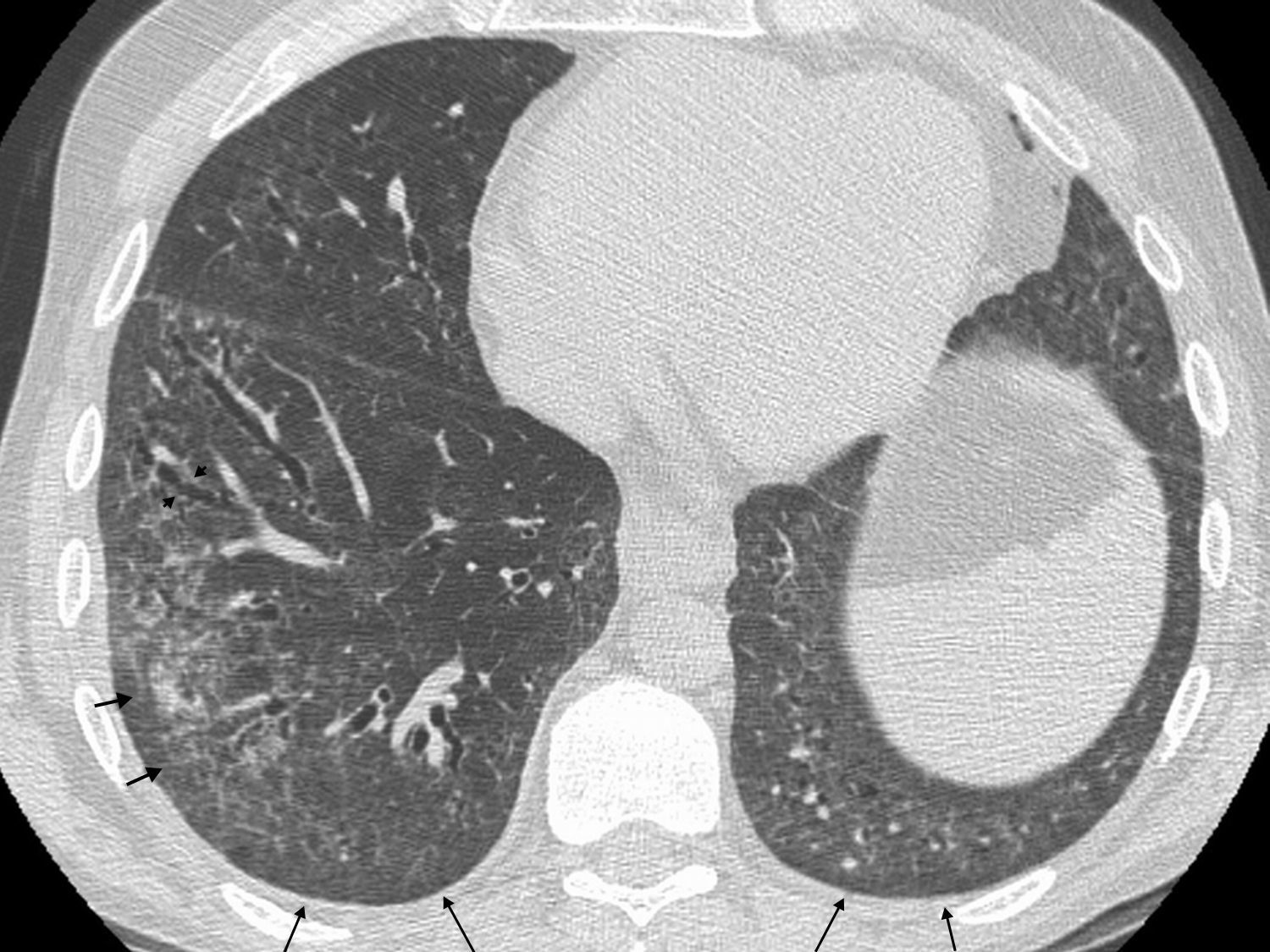
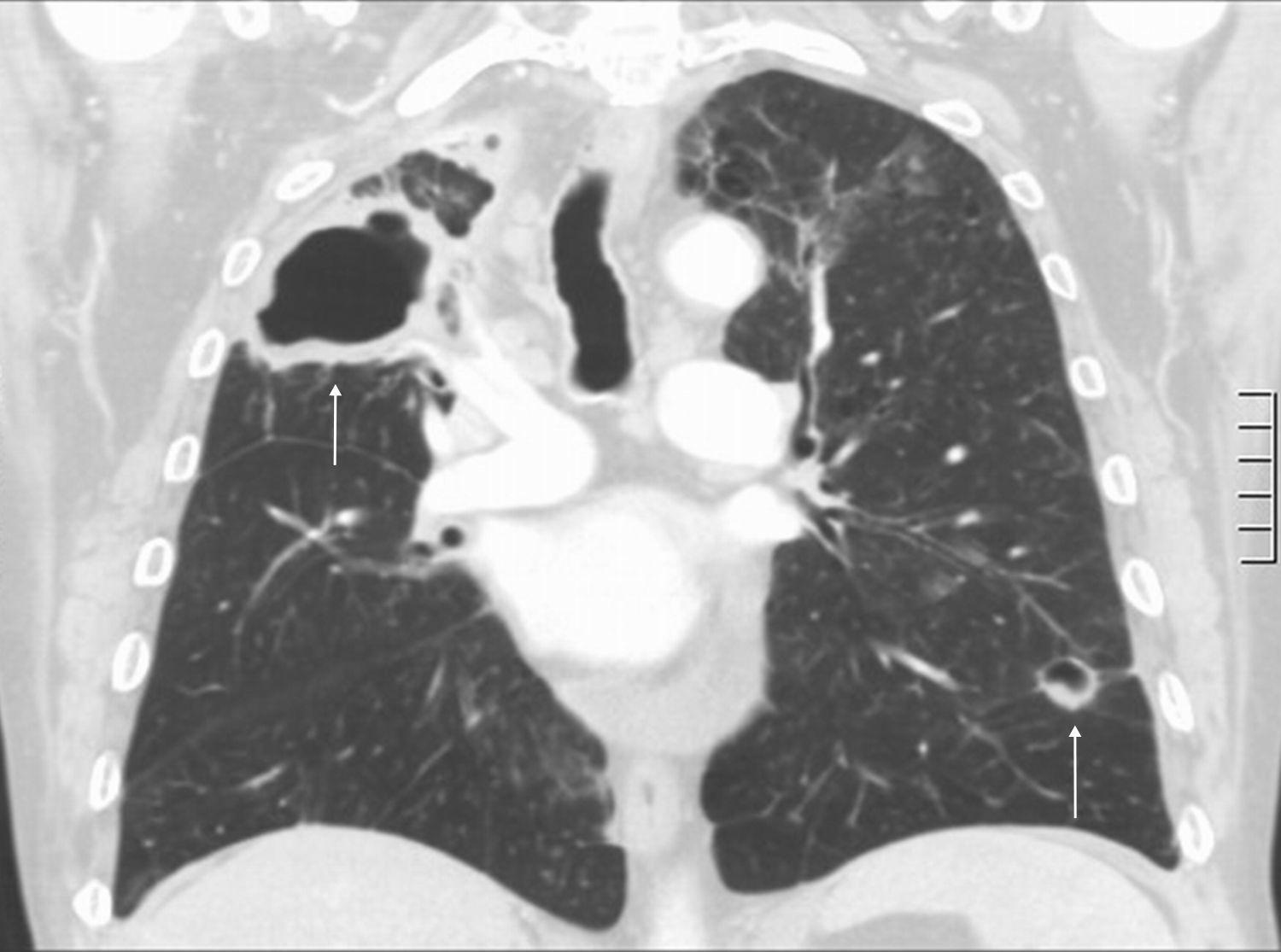
Rheumatoid nodules have been reported in 4% of the patients with AR37 and, as in interstitial lung disease, and they are seen more frequently in men. Nodules are usually associated with the presence of subcutaneous nodules and positive rheumatoid factor, but they can precede the AR manifestations.37 Rheumatoid nodules are usually subpleural, multiple and well-defined, and may often undergo cavitation (Fig. 5). Complications such as pneumothorax and piopneumothorax secondary to rupture of rheumatoid nodules have been reported. Its course varies from spontaneous regression to stabilization.37,45,46
.")
Consolidation is often seen in patients with RA, mainly in the lower zones with a peripheral distribution.5 Presence of consolidation is suggestive of organizing pneumonia,5,24 which in turn may be secondary to drugs or infection. However, this radiologic pattern is unspecific and, when present, a differential diagnosis between organizing pneumonia, infection or acute exacerbation of interstitial pneumonia should be considered.
Pleural abnormalities are a common finding in patients with RA, and pleural thickening is seen more often than pleural effusion on CT.37
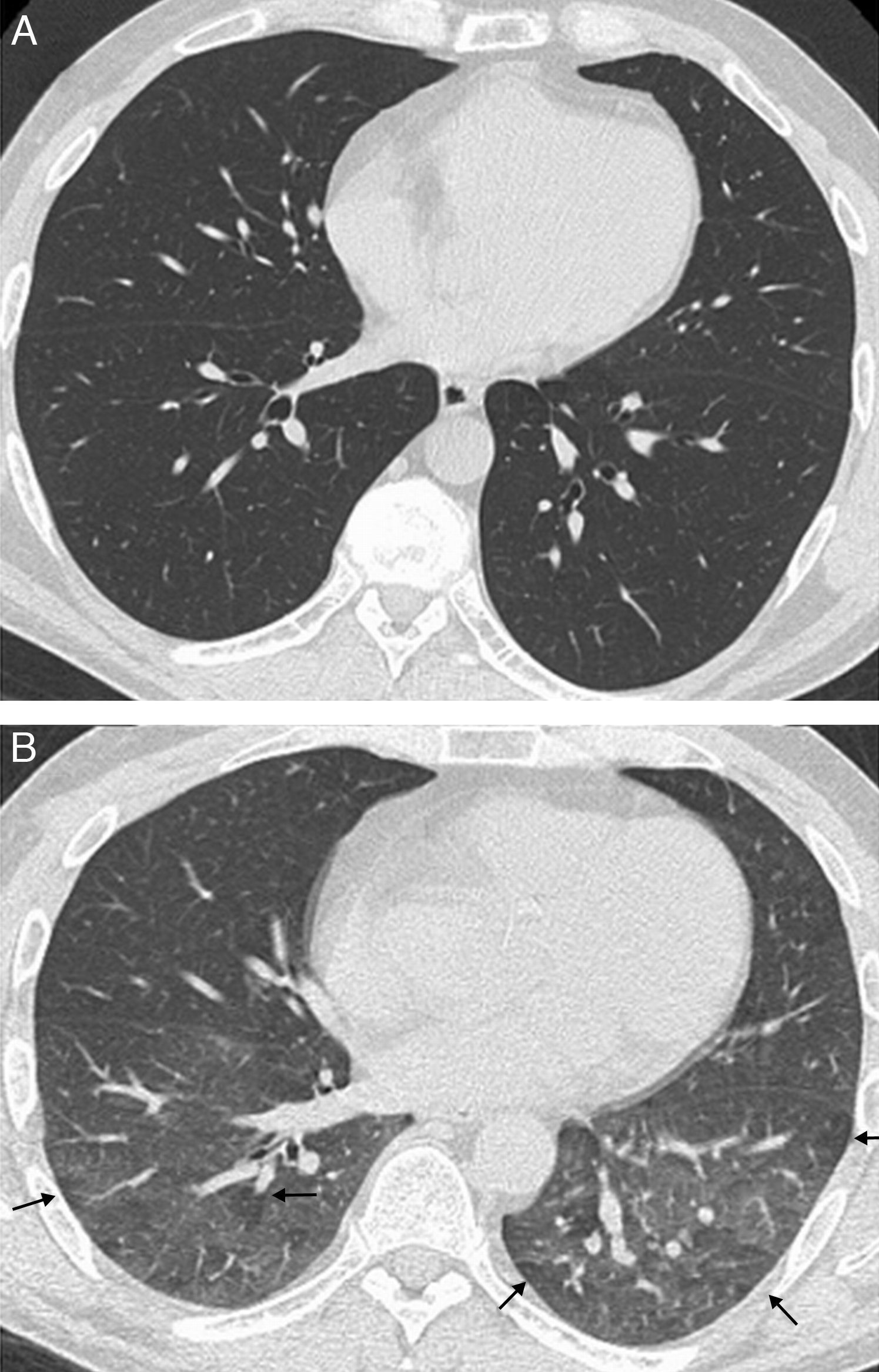
SclerodermaInterstitial lung disease is the most common lung condition in patients with SCL, and the most common cause of death in these patients.2 In SCL, unlike what happens in RA, NSIP is more frequent than UIP,4 which correlates with the typical HRCT findings (ground-glass opacity associated with fine reticulation typically subpleural, basal and posterior) (Fig. 6). The radiologic pattern in SCL is often similar to that of fibrotic NSIP, with traction bronchiectasis and bronchiolectasis (Fig. 7).47,48 Honeycombing is absent or mild, although it has been reported in up to 37% of patients with SCL associated with symptomatic interstitial lung disease.47
 and traction bronchiectasis (white arrows), with basal predominance. Absence of honeycombing.")
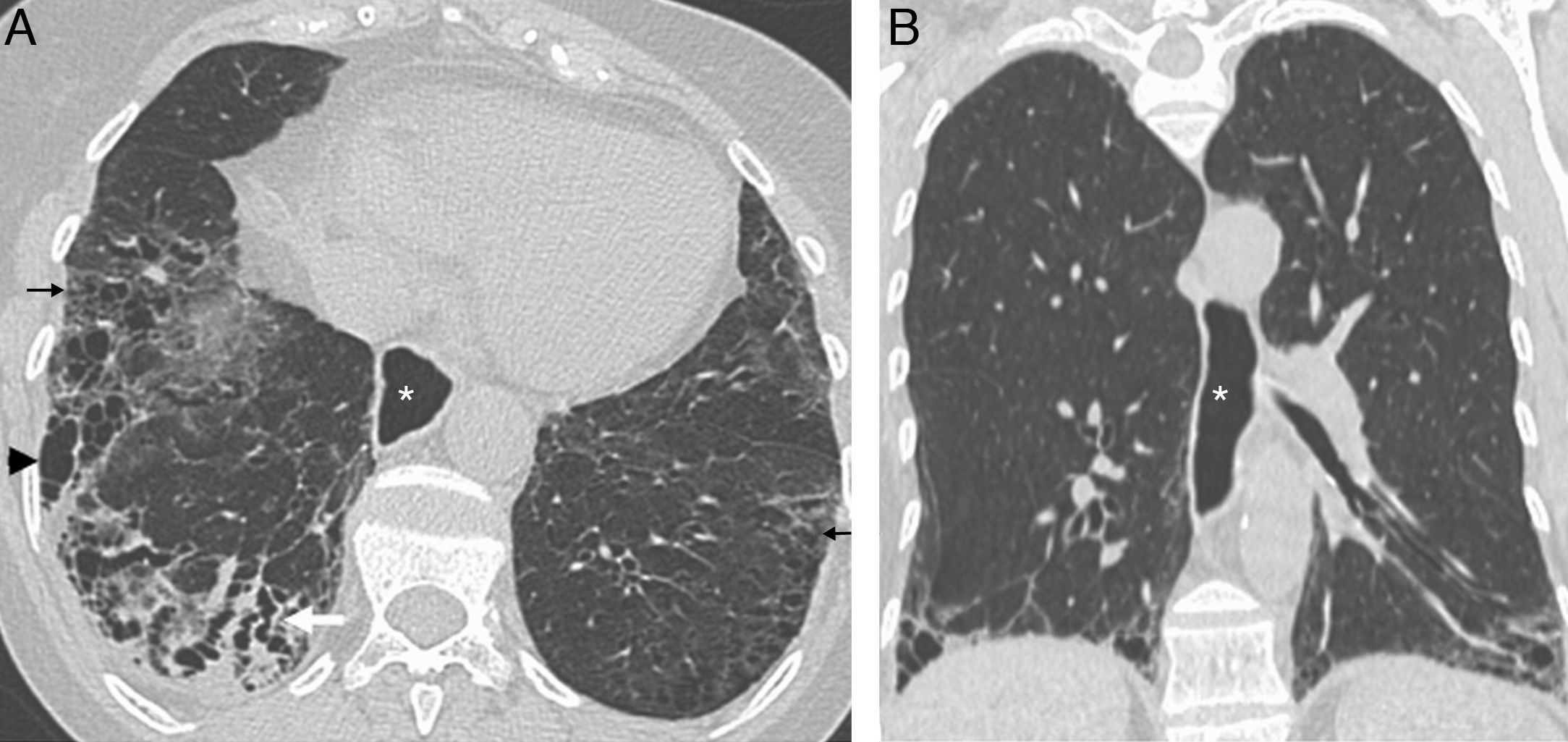
 and coronal (B) HRCT images show a bilateral reticular pattern (arrows), traction bronchiectasis (white arrow) and honeycombing (arrowhead) in RLL. The lesions have a subpleural and basal distribution. The presence of esophageal dilation (*) suggests a diagnosis of scleroderma.")
Systemic scleroderma. Axial (A) and coronal (B) HRCT images show a bilateral reticular pattern (arrows), traction bronchiectasis (white arrow) and honeycombing (arrowhead) in RLL. The lesions have a subpleural and basal distribution. The presence of esophageal dilation (*) suggests a diagnosis of scleroderma.
The presence of honeycombing at diagnosis is correlated with restrictive functional impairment and a worse prognosis.4,47 Tashkin et al.49 reported significant lung function impairment in patients with honeycombing on the baseline HRCT treated with placebo during one year, but this was not observed in patients treated with cyclophosphamide during the same period. However, the positive effect seen in the second group did not persist one year after discontinuing therapy. Pulmonary fibrosis associated with SCL has better prognosis than idiopathic pulmonary fibrosis.50
Pulmonary hypertension (PHT) is particularly common in patients with limited SCL.27,51 Enlargement of the main and proximal pulmonary arteries is seen on CT; however, normal-sized pulmonary arteries do not exclude the diagnosis. Pericardial thickening or effusion also correlates with the presence of echocardiographic pulmonary hypertension.52 Launay et al. compared 47 patients with SCL, PHT and interstitial lung disease with 50 patients with SCL and HTP, but found no interstitial disease. The first group had more frequently diffuse SCL and worse prognosis than the second group. The presence of pericardial effusion was predictive of a poor outcome.53
SCL is associated with a higher risk of lung cancer, regardless of the presence of other risk factors.36
Esophageal abnormalities are common and the presence of esophageal dilation in a patient with interstitial lung disease is suggestive of SCL (Fig. 7). Esophageal dysmotility may cause aspiration pneumonia in these patients.
Systemic lupus erythematosusThe most common pleuropulmonary manifestation of LES is pleuritis (found in 77.8% of necropsies), associated or not with effusion.31 Infection is the most frequent pulmonary complication, and sepsis is a major cause of death in LES patients.31,54 Pulmonary hemorrhage is also an important complication of this condition55 characterized by patchy or diffuse ground-glass opacity or consolidation on CT (Fig. 8A). DAD accounts for 22% of all pulmonary abnormalities in LES and is usually associated with pulmonary hemorrhage, edema or pleural effusion.55 Lupus pneumonia corresponds histologically to varying degrees of hemorrhage and DAD, which manifests as ground-glass opacity and extensive consolidation on CT.54
 Pulmonary hemorrhage in a patient with lupus erythematosus. Axial HRCT image shows bilateral, patchy ground-glass opacities (white arrows). (B) Interstitial lung disease in a patient with lupus erythematosus and restrictive spirometry pattern. Axial HRCT image show ground-glass opacities (arrows), intralobular reticulation and septal lines (circle).")
Systemic lupus erythematosus. (A) Pulmonary hemorrhage in a patient with lupus erythematosus. Axial HRCT image shows bilateral, patchy ground-glass opacities (white arrows). (B) Interstitial lung disease in a patient with lupus erythematosus and restrictive spirometry pattern. Axial HRCT image show ground-glass opacities (arrows), intralobular reticulation and septal lines (circle).
Interstitial lung disease is less common than in the rest of CTDs, but mild HRCT changes are found in one-third of asymptomatic patients, usually ground-glass opacity associated with intralobular reticulation, interlobular septal lines and parenchymal bands56 (Fig. 8B).
Other abnormalities associated with LES include pulmonary hypertension (less common than in SCL), diaphragmatic weakness, and thrombosis secondary to antiphospholipid syndrome, frequently associated with LES.1,54
Polymyositis/dermatomyositisNSIP is the most common pulmonary disease in patients with PM/DM, followed by organizing pneumonia.6,57 Radiologic findings are correlated with histologic findings, being ground-glass opacity, intralobular reticulation, traction bronchiectasis and consolidation the most frequent radiologic findings, with a lower lobe predominance58 (Fig. 2). Consolidation is more commonly seen in PM/DM than in the rest of CTDs, and the histologic pattern corresponds to organizing pneumonia (Fig. 9),6,57,59 with acute pulmonary decompensation with underlying histopathology of DAD (often leading to patient demise)59 and aspiration pneumonia secondary to the muscle disease.1,2 Lung abnormalities may precede muscle and skin lesions in 33% of patients, and there is no relation between the severity of skin/muscle lesions and the appearance of interstitial lung disease.60
; mild subpleural basal reticulation (white circle).")
Female patient with polymyositis and anti-Jo-1-positive antibodies. Radiologic pattern of organizing pneumonia. Axial HRCT images show nodular and lineal ground-glass opacity in both lung bases, with peribronchial and perilobular distribution (arrows); mild subpleural basal reticulation (white circle).
In a series of 28 patients with DM (12 patients) and PM (16 patients), Fujisawa et al.26 concluded that patients with DM had worse response to corticosteroid therapy and worse prognosis than those with PM. DAD was seen in three patients with DM but in none of the PM patients. No differences in NSIP frequency were observed between DM and PM.
It is worth noting that DM/PM and antisynthetase antibody-positive patients frequently have interstitial lung disease (50–70%),1,60 which may precede myositis in 53% of cases.61 The antisynthetase syndrome consists of the presence of antisynthetase antibodies associated with myositis, interstitial disease, Raynaud's phenomenon, joint pain, exanthema on the hands, and fever.62 Anti-Jo-1 is the most commonly identified antisynthetase antibody, but anti-PL-7, -PL-12, -EJ, -OJ, -KS, -Zo, and -YRS are also related to the syndrome.60,61 Conversely, interstitial disease is less commonly found (10%) in DM/PM and anti-Jo-1-negative patients.1,57,62
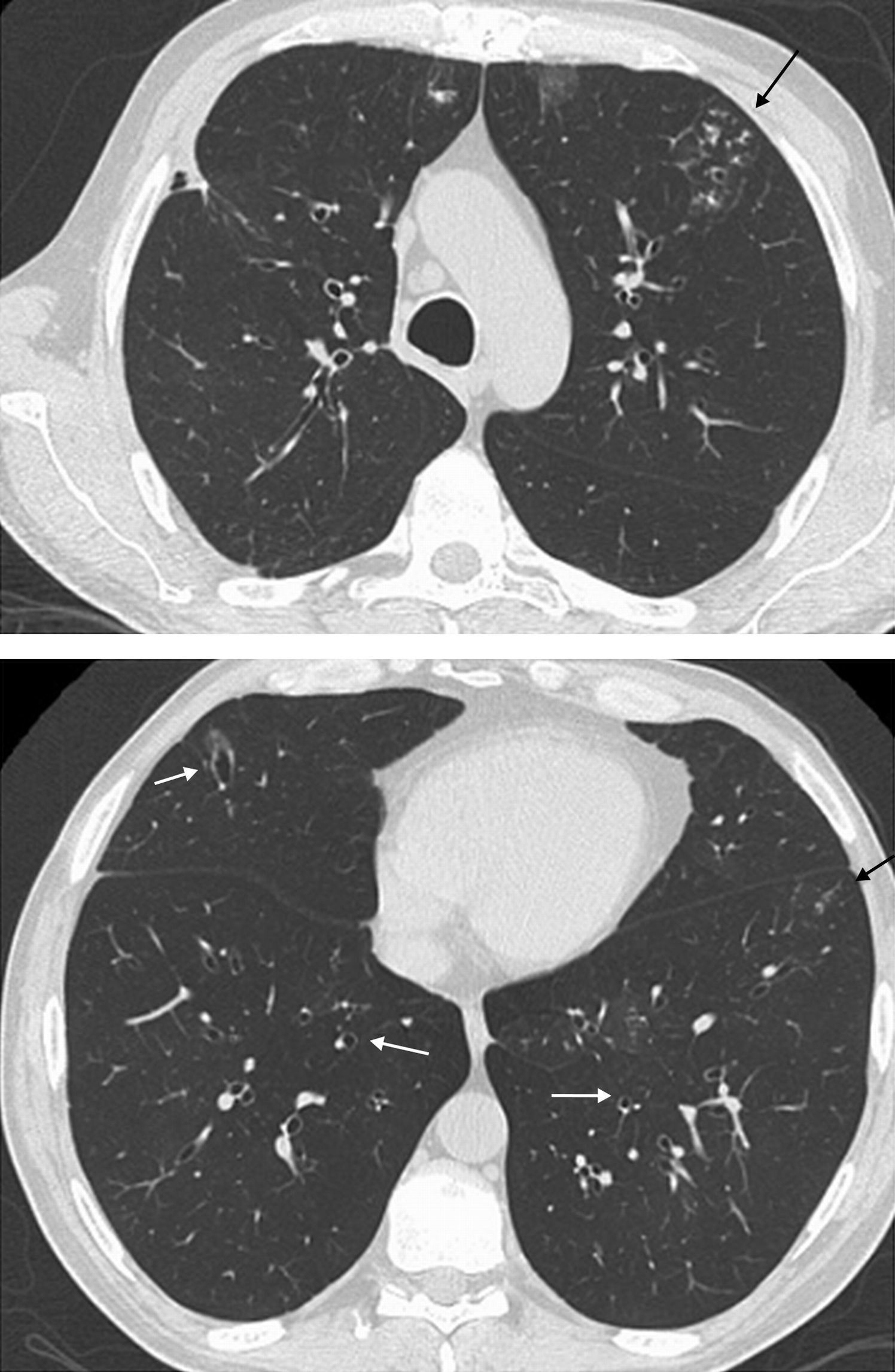
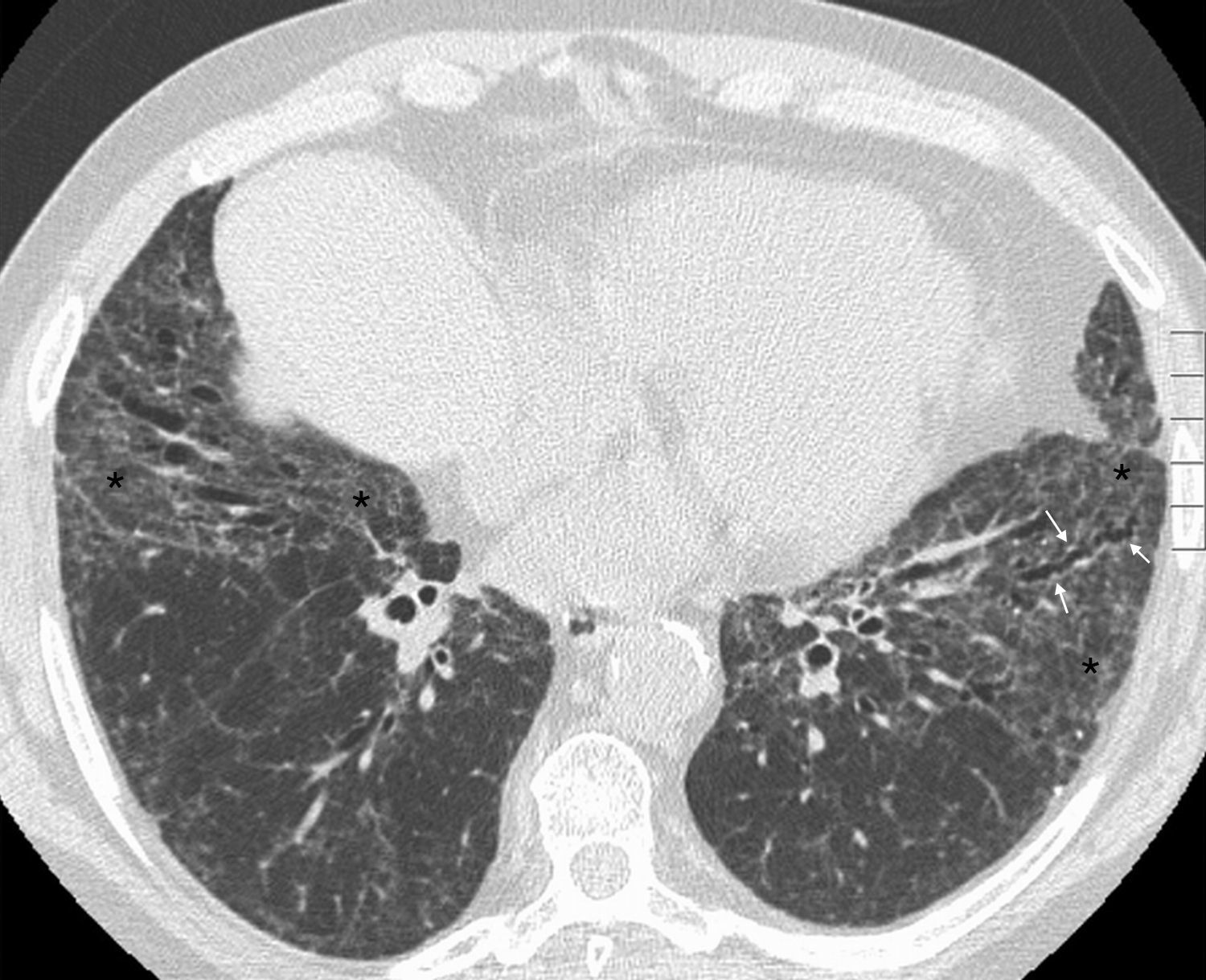
Sjögren's syndromeSjS is relatively common, and it may develop in isolation (primary SjS) or associated with other CTDs, more frequently RA (secondary SjS). The histologic patterns associated with this syndrome include NSIP, bronchiolitis, organizing pneumonia, UIP, LIP, primary pulmonary lymphoma and amyloidosis.4 In two series of 18 and 33 patients with primary SjS and lung biopsy,20,63 NSIP was the most common histologic finding, with good correlation between the histologic and radiologic pattern of NSIP on HRCT (positive predictive value of HRCT 94%).20 Other HRCT findings include bronchiolitis pattern (air trapping, bronchial wall thickening, bronchiectasis/bronchiolectasis and tree-in-bud), peribronchovascular air cysts, nodules, septal lines, and ground-glass opacity (Fig. 10).

Sjögren's syndrome. (A) Patient with primary Sjögren's syndrome and advanced interstitial lung disease. Axial HRCT image of the middle lung field shows micronodules (circles), ground-glass opacity associated with fine reticulation (white arrow) and traction bronchiectasis (black arrow). (B and C) Patient with primary Sjögren's syndrome. Axial HRCT images show perivascular air cysts (arrows).
Air cysts, consolidations, ground-glass opacity, septal lines and nodules have been described in LIP and lymphoma.63,64 Septal lines, nodules and cysts have also been found in amyloidosis.63 Therefore, these findings are not correlated with a specific histologic pattern in SjS, and biopsy is required to typify the disease.20,63 In a series of 50 patients with primary SjS, Franquet et al.65 reported CT abnormalities in 17 patients, being bronchiolar changes and parenchymal lines, followed by ground-glass opacity, the most common findings. The presence of micronodules, tree-in-bud, and honeycombing was also observed. The radiologic findings of SjS generally correspond to a combination of interstitial lung disease and airway disease.
Mixed connective tissue diseaseMixed connective tissue disease (MCTD) is characterized by a combination of LES, SCL and PM/DM findings, and the presence of anti-RNP antibodies. Pulmonary abnormalities are common and similar to those seen in previously described CTDs. In a series of 44 patients with MCTD, Bodolay et al.66 reported active interstitial lung disease in 66% of patients. Ground-glass opacity alone (78%) or in association with mild fibrosis and parenchymal lines (21%) was the most common HRCT finding. The peripheral distribution of lesions with lower lobe predominance is compatible with the radiologic pattern of NSIP. Honeycombing is very rare.66,67 Pulmonary hypertension is present in 4% of patients with MCTD and is a major cause of patient demise, ahead of respiratory failure and heart failure.67 CT may demonstrate an enlarged pulmonary artery. Other associated findings include pleural thickening and effusion27 and esophageal abnormalities.67
ConclusionCTDs are associated with a wide spectrum of lung conditions, which result in significant morbidity and mortality. The radiologist should be familiar with these entities and systematically evaluate the radiologic images in order to diagnose interstitial, airway, pleural or vascular disease, and correlate these findings with the underlying CTD. It should be taken into account that lung changes may precede CTD symptoms and that, in some cases, the radiologic pattern of NSIP, the detection of lung cysts or coexistence of an interstitial and bronchiolar pattern should raise the suspicion of underlying CTD. HRCT plays an important role in the early detection of pulmonary disease in patients with CTD and in the detection of subclinical pulmonary disease in certain situations such as SCL and antisynthetase syndrome, facilitating an early treatment. In addition, the high rate of infections, drug-related adverse reactions and a higher incidence of lung cancer and lymphoma should always be considered in patients with pulmonary disease associated with a CTD.
A multidisciplinary approach involving clinical, radiologic and histologic findings provides a more accurate diagnosis and treatment of these diseases.
Ethical responsibilitiesHuman and animal protection. The authors declare that no experiments involving animals or humans have been carried out for this study.
Data confidentialityThe authors declare that no data from patients are shown in this article.
Right to privacy and informed consentThe authors declare that no data from patients are shown in this article.
Authorship- 1.
Responsible for the integrity of the study: RYM.
- 2.
Conception of the study: RYM.
- 3.
Design of the study: RYM, ABY, SEP, MBN and RRM.
- 4.
Data acquisition: RYM, ABY, SEP and MBN.
- 5.
Analysis and interpretation of data: RYM, ABY, SEP, MBN and RRM.
- 6.
Statistical analysis: N/A.
- 7.
Bibliographic search: RYM, ABY and RRM.
- 8.
Writing of the article: RYM, ABY and RRM.
- 9.
Critical review with intellectually relevant contributions: RYM, ABY, SEP, MBN and RRM.
- 10.
Approval of the final version: RYM, ABY, SEP, MBN and RRM.
The authors declare not having any conflict of interests.
Please cite this article as: Ysamat Marfá R, et al. La patología pulmonar asociada a las enfermedades del tejido conectivo. Radiología. 2013;55:107-17.

