





La hipercolesterolemia familiar homocigótica (HFHo) se caracteriza por niveles muy elevados de cLDL y por enfermedad aterosclerótica temprana. Aunque la frecuencia es baja (1/300.000), las complicaciones son muy severas y pueden ser evitadas. Encontrar y tratar esta población de manera temprana podría reducir la mortalidad. Se describen 36 casos en Colombia, en donde se calcula que haya entre 160 y 200 casos.
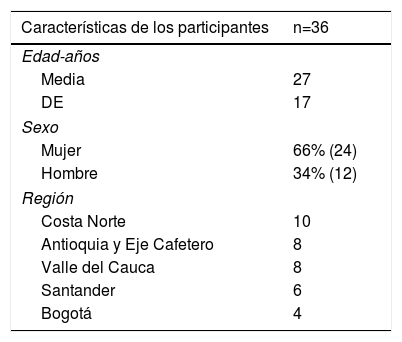
ResultadosUn total de 36 pacientes con fenotipo sugestivo de HFHo fueron identificados y tratados en un período de observación de cuatro años. La media de edad fue 27 años (24 mujeres). 34 pacientes tuvieron un puntaje según la Red de Clínicas de Lípidos de Holanda (RCLH) mayor de 8 (diagnóstico definitivo) y los restantes 2 tenían puntaje equivalente a diagnóstico probable. Un cuarto de los casos procedían de la costa norte colombiana. En las pruebas genéticas, 14 fueron homocigóticos verdaderos para mutación del gen que codifica para el receptor de LDL (LDLR), 12 heterocigóticos compuestos, 2 heterocigóticos dobles y uno autosómico recesivo (LDLRAP1); 5 pacientes fueron heterocigóticos simples (LDLR) y 2 pacientes no autorizaron la prueba. En los homocigóticos verdaderos, la variante más frecuente encontrada fue la c.11G>A. 14 pacientes cursaron con enfermedad coronaria, 9 con estenosis carotídea, 8 con estenosis aórtica y 2 tuvieron ataques cerebrovasculares (ACV). 34 pacientes recibían estatinas (24 rosuvastatina), 30 recibían ezetimibe, 2 recibían evolocumab y 20 recibían lomitapide (dosis promedio 12,7mg). Ninguno recibió aféresis de cLDL. Los medicamentos, en general, fueron bien tolerados y la reducción promedio de cLDL con la terapia fue de 533,7mg/dl a 245,1mg/dl (54%).
ConclusionesTodos los pacientes recibieron tratamiento hipolipemiante y se encontraron alteraciones genéticas diagnósticas en todos aquellos que autorizaron el examen. Los niveles elevados de cLDL conllevan tanto riesgo que el tratamiento debe establecerse aún sin conocer el diagnóstico genético.
Homozygous familial hypercholesterolemia (HoFH) is characterized for very high levels of cLDL and early cardiovascular disease. Although incidence is low (1/300 000), complications are very severe and can be avoided. Finding and treating this population promptly could reduce mortality. We describe 36 cases in Colombia, where 160 to 200 cases are expected.
Results36 patients with phenotype of HoHF were identified and treated in a follow-up of 4 years. The mean age was 27 years (24 women). 34 of them had at least 8 points in the FH Dutch Lipid Clinic Criteria (definitive diagnosis) and two had probable diagnosis. A quarter of the cases came from the Colombian North Coast. In molecular tests, 14 were true homozygous for LDLR, 12 were compound heterozygous for LDLR, 2 double heterozygous and one was autosomal recessive; 5 were heterozygous and 2 patients did not authorized genetic test. In true homozygous subjects, the most frequent variant was c.11G>A. 14 patients had coronary disease, 9 carotid stenosis, 8 aortic stenosis and 2 had stroke. 34 patients were on statins (25 rosuvastatin), 30 were receiving ezetimibe, 2 were receiving a PSCK9 inhibitor (evolocumab) and 20 were on lomitapide with mean doses of 12.7mg. None received lipoprotein apheresis. Medications were very well tolerated. Changes in cLDL after therapy was from 533.7 mg/dL to 245 mg/dL, (54%).
ConclusionsTreatment was started in all patients. We found genetic mutations in all patients with genetic tests. The high levels of cLDL mean such a high risk that treatment must be started promptly, even without a genetic test.
La hipercolesterolemia familiar homocigótica (HFHo) es un trastorno hereditario, casi siempre autosómico dominante, que se caracteriza clínicamente por la presencia de niveles de cLDL muy elevados y enfermedad cardiovascular arteriosclerótica (ECVA) prematura y progresiva; en muchos pacientes pueden encontrarse xantomas o xantelasmas1–3. Las alteraciones genéticas (que deben involucrar los dos alelos) más frecuentes se encuentran en el gen que codifica el receptor para LDL (LDLR) hasta en el 95%, en el gen que codifica la ApoB (APOB) del 2 a 5%, en el gen que codifica la proproteína convertasa de subtilisina/kexina tipo 9 (PCSK9) en menos del 1% y en el gen que codifica la proteína adaptadora 1 del receptor para LDL (LDLRAP1) en menos del 1%4. Otras alteraciones menos frecuentes son las mutaciones para lipasa ácida lisosomal (LIPA) y las alteraciones en genes de ABCG5 y ABCG8, que codifican las esterolinas 1 y 21,5,6. Sin embargo, hasta en 2 a 5% de los sujetos con hipercolesterolemia severa pueden no encontrarse estas mutaciones7,8.
La prevalencia de HFHo es variable según las fuentes consultadas. Aunque inicialmente se estimaba una prevalencia de uno en un millón, algunos estudios recientes han mostrado prevalencias más altas, alrededor de 1 en 300.000 a 500.000 personas9,12. La prevalencia es diferente en diversas regiones del mundo. Un estudio en África reportó una prevalencia de 1 en 30.000 personas10; en Europa, en Dinamarca es de 1 en 160.000 y en España de 1 en 450.000 personas11. En Colombia se estima que puede haber de 160 a 300 casos de HFHo5; un estudio en Medellín estima que podría haber 196.422 casos de HF en Colombia13.
Las manifestaciones clínicas dependen de la actividad residual del LDLR. Además de la elevación de cLDL mayor de 500mg/dl y enfermedad aterosclerótica en edades tan tempranas como desde los dos años; también se pueden encontrar xantomas en piel, tendones y articulaciones, y arco corneal7,14. Se ha demostrado que el diagnóstico y tratamiento oportunos reducen la mortalidad por enfermedad cardiovascular atrosclerótica y mejoran la calidad de vida de estos pacientes15–17.
MétodosEn un período que abarcó desde enero de 2013 hasta diciembre de 2018 se recolectó información de pacientes a través de la Fundación Colombiana para Enfermedades Huérfanas (FUNCOLEHF). Todos los pacientes con alta sospecha de HFHo, diagnosticados y tratados por endocrinólogos, cardiólogos, internistas, pediatras, hepatólogos y genetistas fueron incluidos en el estudio, en total 36 sujetos. En todos los casos (con dos excepciones) se realizó estudio molecular a través de paneles de secuenciación de nueva generación (NGS).
El objetivo general fue describir a 36 pacientes colombianos con fenotipo de HFHo, su distribución geográfica, sexo, edad, puntaje según criterios RCLH, niveles de colesterol, características clínicas y tratamientos recibidos, así como efectos adversos a la terapia, al igual que describir los resultados de las pruebas genéticas, las mutaciones encontradas, los eventos no fatales relacionados con enfermedad cardiovascular, como estenosis aórtica o carotídea, ataque cererovascular y enfermedad coronaria, y los niveles de colesterol total (CT), cLDL, cHDL y triglicéridos (Tg) basales y el último resultado disponible durante el seguimiento y tratamiento. Los datos se recolectaron y almacenaron en Microsoft Excel con precaución para preservar la confidencialidad.
Tanto los pacientes como los médicos tratantes aceptaron la participación en este estudio y autorizaron a la fundación FUNCOLEHF para la recolección y análisis de sus datos; también aceptaron la publicación de esta serie de casos. Se incluyó a pacientes con cLDL mayor de 300mg/dl si recibían hipolipemiantes en dosis máximas toleradas, o cLDL mayor de 500mg/dl si no recibían tratamiento. El análisis de pruebas genéticas fue realizado por un experto en genética clínica (HP). A todos los pacientes se les realizaron pruebas de función hepática durante el curso de tratamiento con hipolipemiantes y se documentaron los efectos adversos presentados con estas terapias.
Análisis estadísticoPara el análisis se hicieron descripciones de las variables utilizando medias, medianas, rangos y desviaciones estándar, y las frecuencias en porcentajes. Para el análisis estadístico se utilizó el programa Stata (StataCorp) versión 15.0 para Mac.
ResultadosLa edad promedio de los 36 pacientes fue 27 ± 17 años; 24 fueron mujeres. La distribución demográfica y geográfica se encuentra en la tabla 1.
El signo clínico más encontrado fueron los xantomas tendinosos, en 26 pacientes; en 19 se encontró arco corneal y en 7 había xantelasmas. 20 pacientes cursaron con ECVA, con enfermedad coronaria como manifestación más frecuente (n=14), seguida de estenosis aórtica y carotídea en 9 y 8, respectivamente. Dos pacientes tuvieron ataque cerebrovascular.
La mayoría de los pacientes tenían un diagnóstico definitivo de HF según la RCLH. Los datos completos están en la tabla 2.
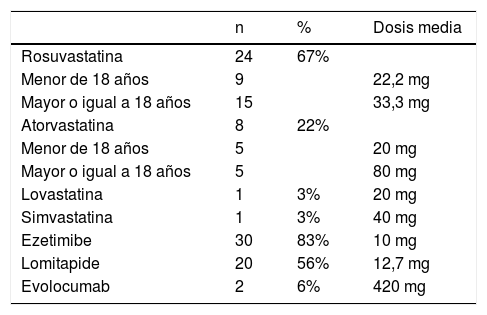
TratamientoDel total de pacientes, 34 recibieron estatinas, de las cuales la más frecuente (n=24) fue rosuvastatina, en dosis promedio de 22,2mg en pacientes menores de 18 años (edad mínima de 4 años) y de 33,3mg en pacientes mayores de 18 años. La segunda terapia más usada fue ezetimibe, en 30 pacientes, todos con 10mg. En 20 pacientes se prescribió lomitapide, a una dosis promedio de 12,7mg; a dos pacientes se les agregó evolocumab (tabla 3). Los efectos adversos más comunes fueron molestias gastrointestinales leves en 6 pacientes, y elevación de las transaminasas hasta dos veces el límite superior en 5 pacientes; en un paciente hubo elevación de CPK, razón por la cual los médicos tratantes disminuyeron la dosis de estatina (tabla 4).
Tratamiento hipolipemiante
| n | % | Dosis media | |
|---|---|---|---|
| Rosuvastatina | 24 | 67% | |
| Menor de 18 años | 9 | 22,2 mg | |
| Mayor o igual a 18 años | 15 | 33,3 mg | |
| Atorvastatina | 8 | 22% | |
| Menor de 18 años | 5 | 20 mg | |
| Mayor o igual a 18 años | 5 | 80 mg | |
| Lovastatina | 1 | 3% | 20 mg |
| Simvastatina | 1 | 3% | 40 mg |
| Ezetimibe | 30 | 83% | 10 mg |
| Lomitapide | 20 | 56% | 12,7 mg |
| Evolocumab | 2 | 6% | 420 mg |
Valores basales y finales de lípidos, y cambio en porcentaje
| Niveles basales | Niveles finales | Cambio | |
|---|---|---|---|
| TC (mg/dl) | 607,3 | 305,9 | 50% |
| LDL-C (mg/dl) | 533,7 | 245,1 | 54% |
| Estatina más ezetimibe sin lomitapide (n=11) | 487,4 | 319,1 | 35% |
| Estatina más ezetimibe más lomitapide (n=18) | 602,0 | 190,5 | 68% |
| Estatina y ezetimibe, o ambas, más evolucumab (n=2) | 439,6 | 413,5 | 5% |
| Estatina sola (n=1) | 299 | 183 | 38% |
| HDL (mg/dl) | 40,7 | 47,7 | 17% |
| TG (mg/dl) | 142,4 | 115,4 | 19% |
A consideración de cada médico tratante, hubo 29 pacientes con estatina y ezetimibe. En el grupo de menores de 7 años (n=4), se inició con dosis menores de estatina en monoterapia (n=1) o ezetimibe solo (n=1) y dos pacientes con estatina e iPCSK9 sin ezetimibe.
En el reclutamiento el promedio de cLDL basal fue de 533,7mg/dl. En la tabla 2 se muestran los lípidos basales y del último perfil, luego de tratamiento, así como el porcentaje de cambio en promedio. La misma tabla muestra los cambios en el perfil lipídico después del tratamiento. Los niveles iniciales en promedio de colesterol total y LDL fueron 607mg/dl y 533mg/dl respectivamente. Después de la terapia hipolipemiante hubo una disminución de aproximadamente 50% en estos dos últimos valores. Adicionalmente se evidenció un cambio en triglicéridos y HDL de 17 y 19%, respectivamente. Ninguno de los pacientes recibió terapia con aféresis de cLDL, la cual no está disponible en Colombia.
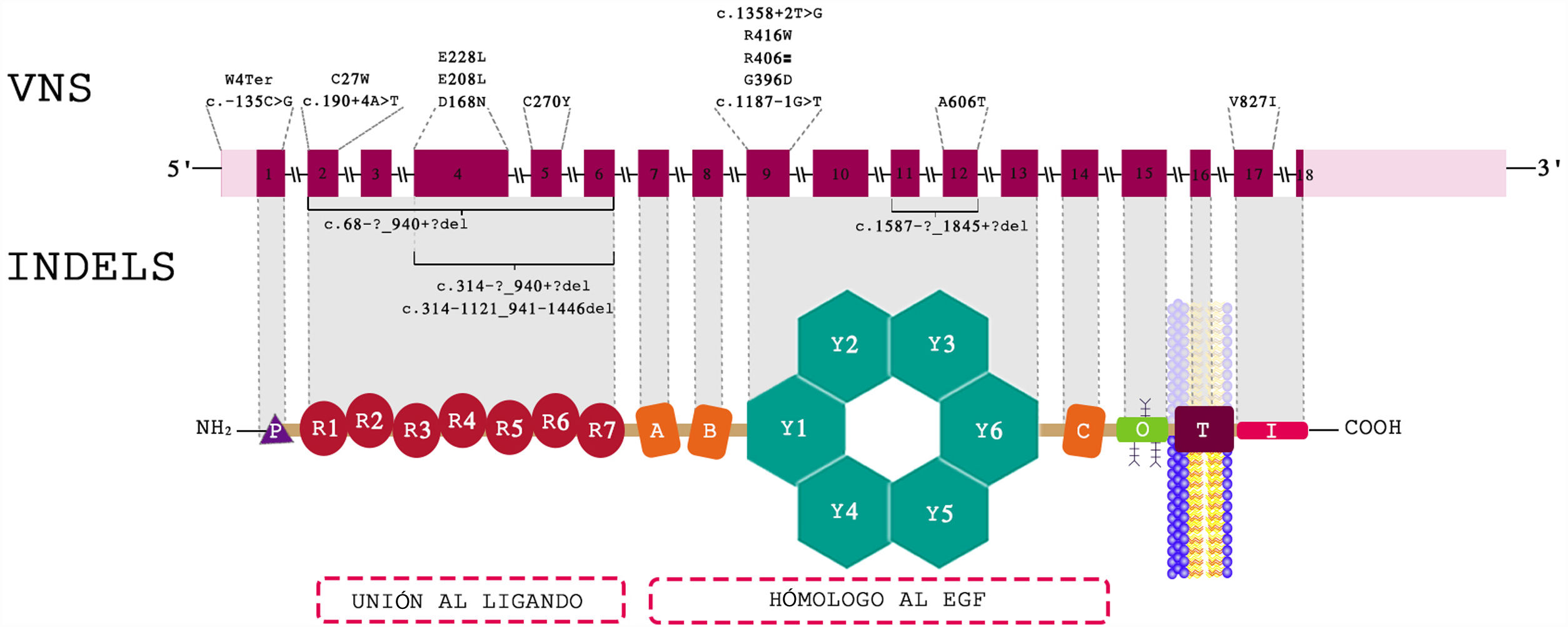
Hallazgos genéticosLa variante más frecuente fue la encontrada en el primer exón del gen LDLR, que se halla en el brazo corto del cromosoma 19, c.11G>A tipo nonsense, que produce un codón de parada que genera una proteína truncada (p.Trp4Ter). En la figura 1 aparece la distribución de las variantes a lo largo del gen LDLR.

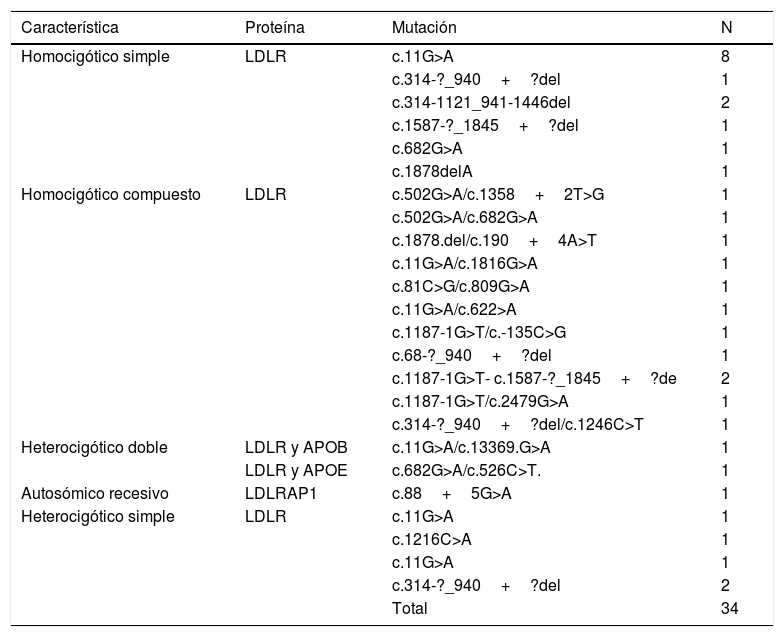
14 de los pacientes fueron homocigóticos simples o verdaderos, con la mutación c.11G>A clasificada como patogénica en ambos alelos en 8 pacientes, todos provenientes de la Costa Atlántica. Se encontraron 12 casos heterocigóticos compuestos, todos codificaban LDLR, 2 casos de heterocigóticos dobles para LDLR/APOB y LDLR/APOE, y 1 caso autosómico recesivo para LDLRAP1. Los 5 casos restantes fueron heterocigóticos simples por genotipo, pero con comportamiento clínico como homocigóticos verdaderos, con puntuación de RCLH mayor de 8.
La tabla 5 muestra la clasificación según genotipos, sexo y sitios de procedencia, en tanto que en la tabla 6 se muestran las mutaciones encontradas en todos los pacientes.
| Hallazgos genéticos | n=34% (n) |
|---|---|
| Homocigótico simple | 38,9% (14) |
| Edad (media) | 22,5 |
| Mujeres | 42%(6) |
| Región Costa Norte | 50%(7) |
| Heterocigótico compuesto | 33,3% (12) |
| Edad (media) | 24,2 |
| Mujeres | 66,6 (8) |
| Región Santander | 25% (3) |
| Región Eje Cafetero | 25% (3) |
| Heterocigótico doble | 5,6% (2) |
| Edad (media) | 44,5 |
| Hombres | 100% (2) |
| Región Antioquia | 50% (1) |
| Autosómico recesivo | 2,8% (1) |
| Edad (media) | 8 |
| Hombres | 100% (1) |
| Región Antioquia | 100% (1) |
| Heterocigótico simple | 13,9% (5) |
| Edad (media) | 29,4 |
| Hombres | 100% (1) |
| Región Valle | 40% (2) |
| Característica | Proteína | Mutación | N |
|---|---|---|---|
| Homocigótico simple | LDLR | c.11G>A | 8 |
| c.314-?_940+?del | 1 | ||
| c.314-1121_941-1446del | 2 | ||
| c.1587-?_1845+?del | 1 | ||
| c.682G>A | 1 | ||
| c.1878delA | 1 | ||
| Homocigótico compuesto | LDLR | c.502G>A/c.1358+2T>G | 1 |
| c.502G>A/c.682G>A | 1 | ||
| c.1878.del/c.190+4A>T | 1 | ||
| c.11G>A/c.1816G>A | 1 | ||
| c.81C>G/c.809G>A | 1 | ||
| c.11G>A/c.622>A | 1 | ||
| c.1187-1G>T/c.-135C>G | 1 | ||
| c.68-?_940+?del | 1 | ||
| c.1187-1G>T- c.1587-?_1845+?de | 2 | ||
| c.1187-1G>T/c.2479G>A | 1 | ||
| c.314-?_940+?del/c.1246C>T | 1 | ||
| Heterocigótico doble | LDLR y APOB | c.11G>A/c.13369.G>A | 1 |
| LDLR y APOE | c.682G>A/c.526C>T. | 1 | |
| Autosómico recesivo | LDLRAP1 | c.88+5G>A | 1 |
| Heterocigótico simple | LDLR | c.11G>A | 1 |
| c.1216C>A | 1 | ||
| c.11G>A | 1 | ||
| c.314-?_940+?del | 2 | ||
| Total | 34 |
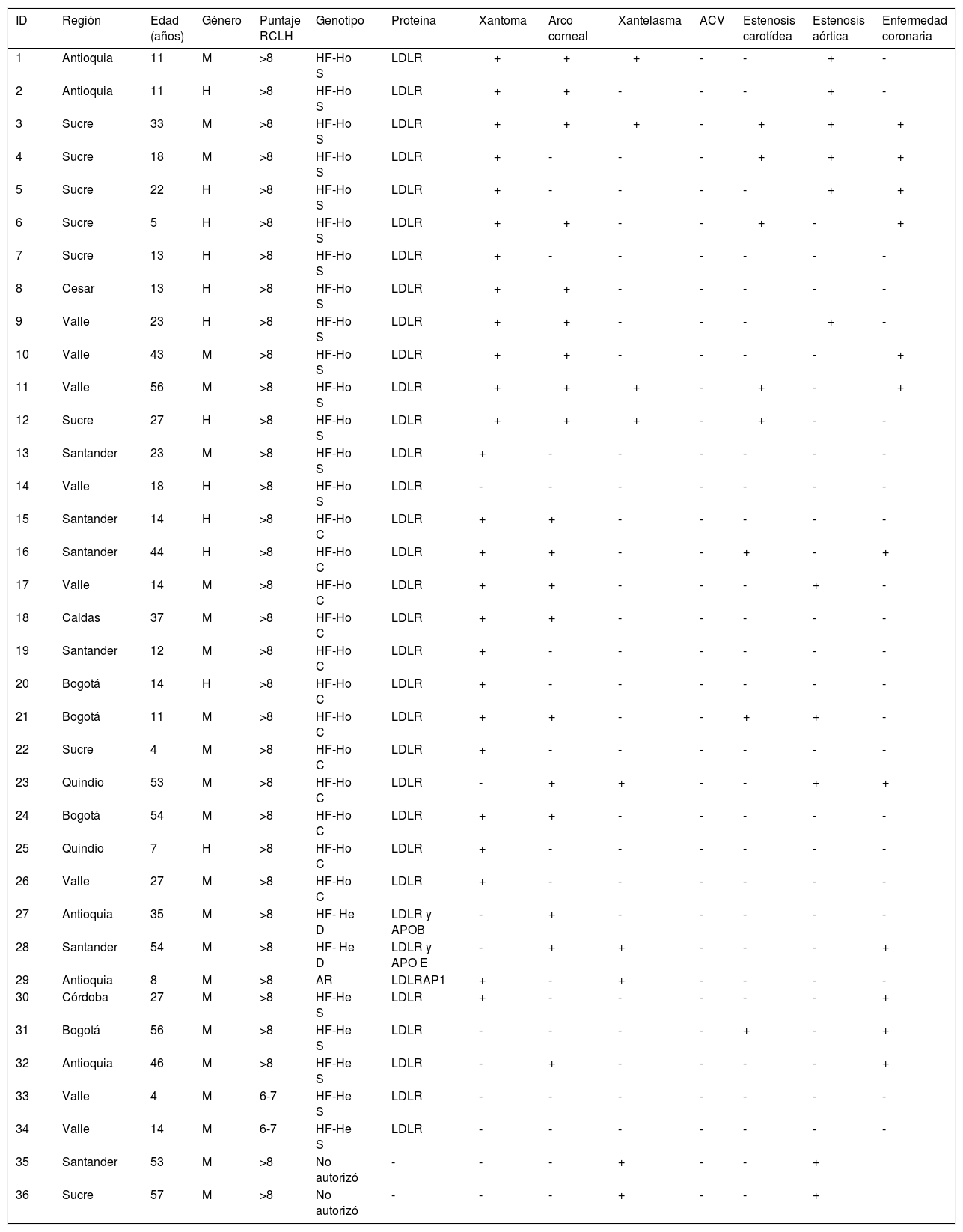
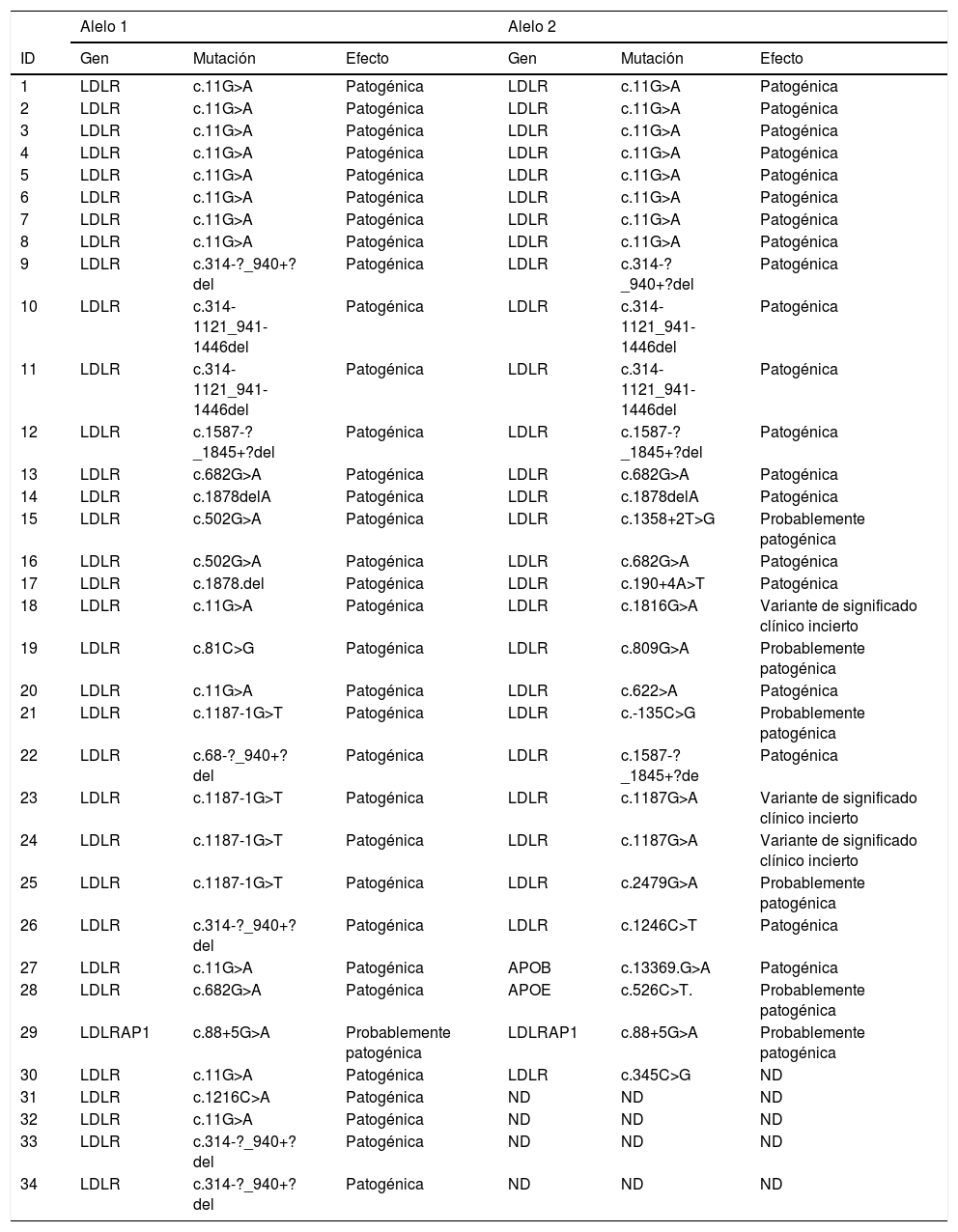
En las tablas 7 y 8 se detallan las características demográficas, clínicas y genéticas de cada paciente.
Características demográficas y clínicas de los pacientes con HF-Ho
| ID | Región | Edad (años) | Género | Puntaje RCLH | Genotipo | Proteína | Xantoma | Arco corneal | Xantelasma | ACV | Estenosis carotídea | Estenosis aórtica | Enfermedad coronaria |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Antioquia | 11 | M | >8 | HF-Ho S | LDLR | + | + | + | - | - | + | - |
| 2 | Antioquia | 11 | H | >8 | HF-Ho S | LDLR | + | + | - | - | - | + | - |
| 3 | Sucre | 33 | M | >8 | HF-Ho S | LDLR | + | + | + | - | + | + | + |
| 4 | Sucre | 18 | M | >8 | HF-Ho S | LDLR | + | - | - | - | + | + | + |
| 5 | Sucre | 22 | H | >8 | HF-Ho S | LDLR | + | - | - | - | - | + | + |
| 6 | Sucre | 5 | H | >8 | HF-Ho S | LDLR | + | + | - | - | + | - | + |
| 7 | Sucre | 13 | H | >8 | HF-Ho S | LDLR | + | - | - | - | - | - | - |
| 8 | Cesar | 13 | H | >8 | HF-Ho S | LDLR | + | + | - | - | - | - | - |
| 9 | Valle | 23 | H | >8 | HF-Ho S | LDLR | + | + | - | - | - | + | - |
| 10 | Valle | 43 | M | >8 | HF-Ho S | LDLR | + | + | - | - | - | - | + |
| 11 | Valle | 56 | M | >8 | HF-Ho S | LDLR | + | + | + | - | + | - | + |
| 12 | Sucre | 27 | H | >8 | HF-Ho S | LDLR | + | + | + | - | + | - | - |
| 13 | Santander | 23 | M | >8 | HF-Ho S | LDLR | + | - | - | - | - | - | - |
| 14 | Valle | 18 | H | >8 | HF-Ho S | LDLR | - | - | - | - | - | - | - |
| 15 | Santander | 14 | H | >8 | HF-Ho C | LDLR | + | + | - | - | - | - | - |
| 16 | Santander | 44 | H | >8 | HF-Ho C | LDLR | + | + | - | - | + | - | + |
| 17 | Valle | 14 | M | >8 | HF-Ho C | LDLR | + | + | - | - | - | + | - |
| 18 | Caldas | 37 | M | >8 | HF-Ho C | LDLR | + | + | - | - | - | - | - |
| 19 | Santander | 12 | M | >8 | HF-Ho C | LDLR | + | - | - | - | - | - | - |
| 20 | Bogotá | 14 | H | >8 | HF-Ho C | LDLR | + | - | - | - | - | - | - |
| 21 | Bogotá | 11 | M | >8 | HF-Ho C | LDLR | + | + | - | - | + | + | - |
| 22 | Sucre | 4 | M | >8 | HF-Ho C | LDLR | + | - | - | - | - | - | - |
| 23 | Quindío | 53 | M | >8 | HF-Ho C | LDLR | - | + | + | - | - | + | + |
| 24 | Bogotá | 54 | M | >8 | HF-Ho C | LDLR | + | + | - | - | - | - | - |
| 25 | Quindío | 7 | H | >8 | HF-Ho C | LDLR | + | - | - | - | - | - | - |
| 26 | Valle | 27 | M | >8 | HF-Ho C | LDLR | + | - | - | - | - | - | - |
| 27 | Antioquia | 35 | M | >8 | HF- He D | LDLR y APOB | - | + | - | - | - | - | - |
| 28 | Santander | 54 | M | >8 | HF- He D | LDLR y APO E | - | + | + | - | - | - | + |
| 29 | Antioquia | 8 | M | >8 | AR | LDLRAP1 | + | - | + | - | - | - | - |
| 30 | Córdoba | 27 | M | >8 | HF-He S | LDLR | + | - | - | - | - | - | + |
| 31 | Bogotá | 56 | M | >8 | HF-He S | LDLR | - | - | - | - | + | - | + |
| 32 | Antioquia | 46 | M | >8 | HF-He S | LDLR | - | + | - | - | - | - | + |
| 33 | Valle | 4 | M | 6-7 | HF-He S | LDLR | - | - | - | - | - | - | - |
| 34 | Valle | 14 | M | 6-7 | HF-He S | LDLR | - | - | - | - | - | - | - |
| 35 | Santander | 53 | M | >8 | No autorizó | - | - | - | + | - | - | + | |
| 36 | Sucre | 57 | M | >8 | No autorizó | - | - | - | + | - | - | + |
HF-Ho S: homocigótico simple, HF-Ho C: heterocigoto compuesto, HF-Ho D: heterocigótico doble. HF-He S: heterocigótico simple.
Genotipificación de los alelos de los pacientes con HF-Ho
| Alelo 1 | Alelo 2 | |||||
|---|---|---|---|---|---|---|
| ID | Gen | Mutación | Efecto | Gen | Mutación | Efecto |
| 1 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 2 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 3 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 4 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 5 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 6 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 7 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 8 | LDLR | c.11G>A | Patogénica | LDLR | c.11G>A | Patogénica |
| 9 | LDLR | c.314-?_940+?del | Patogénica | LDLR | c.314-?_940+?del | Patogénica |
| 10 | LDLR | c.314-1121_941-1446del | Patogénica | LDLR | c.314-1121_941-1446del | Patogénica |
| 11 | LDLR | c.314-1121_941-1446del | Patogénica | LDLR | c.314-1121_941-1446del | Patogénica |
| 12 | LDLR | c.1587-?_1845+?del | Patogénica | LDLR | c.1587-?_1845+?del | Patogénica |
| 13 | LDLR | c.682G>A | Patogénica | LDLR | c.682G>A | Patogénica |
| 14 | LDLR | c.1878delA | Patogénica | LDLR | c.1878delA | Patogénica |
| 15 | LDLR | c.502G>A | Patogénica | LDLR | c.1358+2T>G | Probablemente patogénica |
| 16 | LDLR | c.502G>A | Patogénica | LDLR | c.682G>A | Patogénica |
| 17 | LDLR | c.1878.del | Patogénica | LDLR | c.190+4A>T | Patogénica |
| 18 | LDLR | c.11G>A | Patogénica | LDLR | c.1816G>A | Variante de significado clínico incierto |
| 19 | LDLR | c.81C>G | Patogénica | LDLR | c.809G>A | Probablemente patogénica |
| 20 | LDLR | c.11G>A | Patogénica | LDLR | c.622>A | Patogénica |
| 21 | LDLR | c.1187-1G>T | Patogénica | LDLR | c.-135C>G | Probablemente patogénica |
| 22 | LDLR | c.68-?_940+?del | Patogénica | LDLR | c.1587-?_1845+?de | Patogénica |
| 23 | LDLR | c.1187-1G>T | Patogénica | LDLR | c.1187G>A | Variante de significado clínico incierto |
| 24 | LDLR | c.1187-1G>T | Patogénica | LDLR | c.1187G>A | Variante de significado clínico incierto |
| 25 | LDLR | c.1187-1G>T | Patogénica | LDLR | c.2479G>A | Probablemente patogénica |
| 26 | LDLR | c.314-?_940+?del | Patogénica | LDLR | c.1246C>T | Patogénica |
| 27 | LDLR | c.11G>A | Patogénica | APOB | c.13369.G>A | Patogénica |
| 28 | LDLR | c.682G>A | Patogénica | APOE | c.526C>T. | Probablemente patogénica |
| 29 | LDLRAP1 | c.88+5G>A | Probablemente patogénica | LDLRAP1 | c.88+5G>A | Probablemente patogénica |
| 30 | LDLR | c.11G>A | Patogénica | LDLR | c.345C>G | ND |
| 31 | LDLR | c.1216C>A | Patogénica | ND | ND | ND |
| 32 | LDLR | c.11G>A | Patogénica | ND | ND | ND |
| 33 | LDLR | c.314-?_940+?del | Patogénica | ND | ND | ND |
| 34 | LDLR | c.314-?_940+?del | Patogénica | ND | ND | ND |
ND: no definida.
La recolección de estos casos colombianos se fundamentó en la sospecha clínica de hipercolesterolemia familiar según los criterios de RCLH como escala de probabilidad de certeza y posterior confirmación genética en 34 de los mismos, a diferencia de lo reportado en estudios previos, en los que los casos se describen a partir de bases de datos de estudios genéticos18. La ventaja de usar los criterios RCLH radica en poder relacionar la puntuación de la escala con la probabilidad de encontrar una mutación patogénica19.
La mutación homocigótica c.11G>A, la más encontrada en el actual estudio, no se halló en los estudios de Palacios en España ni en el estudio italiano de D’Erasmo ni el argentino de Gómez9,11,20, pero sí en México y Colombia21,22. Esto sugeriría que cada región geográfica puede tener mutaciones específicas, como se ha descrito en poblaciones con HF23,24. El hallazgo aquí descrito de un caso autosómico recesivo es relevante porque es de baja aparición en población latinoamericana, en tanto que sí es frecuente en población italiana11.
El riesgo cardiovascular varía entre personas con hipercolesterolemia severa, en quienes se ha encontrado que niveles de cLDL mayores a 190mg/dl pueden aumentar el riesgo de ECVA de 6 a 22 veces si la mutación de los genes de HF está presente7. Más de la mitad de los pacientes de esta serie cursaron con alguna enfermedad cardiovascular, lo que contrasta con lo reportado en otros estudios9,11.
Respecto al tratamiento, en otras cohortes se ha observado alta variabilidad en la reducción del cLDL, entre 13 y 97%9. En este estudio la reducción fue del 50%. Un porcentaje significativo de pacientes de este estudio recibían lomitapide, medicamento que históricamente ha sido usado para el manejo de la HFHo9,25, porque su efecto no depende de la presencia o funcionalidad de los receptores para cLDL. En el estudio la dosis promedio de lomitapide era baja, lo que puede explicarse por el alto costo del medicamento y la dificultad para su recobro o por temor a efectos adversos, entre otras razones, como ha pasado en otros casos en Colombia20.
En la cohorte italiana9, el 66% recibió terapia con aféresis de LDL, que se ha descrito como una terapia de rescate que logra disminuir la enfermedad cardiovascular2; no obstante, en Colombia no está disponible. En cuanto al uso de inhibidores de la PCSK9, en dos pacientes se usó evolocumab; sin embargo, no se encontró respuesta positiva porque se trataba de pacientes homocigóticos verdaderos.
Por la alta variabilidad en el manejo y metas de la HF se considera pertinente que los pacientes con alta sospecha clínica de la enfermedad sean derivados de manera temprana a centros de manejo especializado, con miras a reducir el impacto cardiovascular que esta genera. De igual forma, se hace relevante la creación de un programa de tamizado en Colombia que tenga en cuenta la epidemiología local y la baja correlación genotipo-fenotipo descrita. Se ha propuesto que el ideal para la realización de pruebas moleculares y funcionales sea en edades tempranas, idealmente antes de los 40 años26,27.
Esta serie de casos presenta las mutaciones de una muestra de pacientes colombianos, información que resulta útil a la hora de implementar programas de tamizado molecular mediante plataformas de pruebas genéticas basadas en el conocimiento de las mutaciones locales, en el contexto del espectro clínico y molecular de la HF.
Una limitación de este estudio es que el tamaño de muestra no permite extrapolar ni generalizar los resultados a toda la población colombiana; sin embargo, es el primer estudio de caracterización molecular de HF en el país. Por lo tanto, se propone la creación de un registro de HF para Colombia, con base en el modelo del registro SAFEHEART28. Se resalta la importancia de emplear programas de tamizado entre los 9 y 11 años de edad, como ocurre en otros países1,4,14,29.
ConclusionesLa HFHo es una enfermedad infrecuente, sin embargo, con una alta carga de enfermedad cardiovascular desde edades tempranas. La evaluación genética es importante y debería implementarse, no obstante, sin que esto retrase el inicio de tratamiento. La utilidad de esta caracterización molecular en pacientes colombianos servirá como una base para plantear un abordaje multidisciplinario, ampliar el panorama para creación de registros nacionales y estimular más investigación en este campo.
La sospecha clínica debe llevar al diagnóstico, que, a su vez, permite establecer el manejo integral y oportuno. En algunos casos puede ser difícil llegar a la caracterización genotípica por las dificultades logísticas o económicas para la realización de las pruebas. En ningún caso debe retrasarse el tratamiento farmacológico por esperar la caracterización molecular, puesto que debe tratarse el fenotipo, no el genotipo.
Conflictos de interesesNinguno.
Reconocimientos y agradecimientosA la Fundación Colombiana para Enfermedades Huérfanas —FUNCOLEHF—, que a lo largo de los cuatro años ayudó a recolectar esta cantidad importante de pacientes.