


La taquicardia ventricular polimórfica catecolaminérgica es una enfermedad caracterizada por arritmias ventriculares desencadenadas por estrés o actividad física. Existen casos descritos de taquicardia ventricular polimórfica catecolaminérgica asociada a ventrículo izquierdo no compactado, en relación con mutaciones del gen RYR2 localizadas en el exón 3. Se expone el caso clínico de una paciente joven que debutó con clínica de síncopes recurrentes asociados a estrés físico o emocional. En el estudio posterior se descubrió taquicardia ventricular polimórfica catecolaminérgica, con áreas de miocardio no compactado y una nueva variante genética posiblemente asociada a la enfermedad.
Catecholaminergic polymorphic ventricular tachycardia is disease characterised by ventricular arrhythmias triggered by stress or physical activity. There are some cases of catecholaminergic polymorphic ventricular tachycardia described that are associated with non-compacted left ventricle in relation to mutations of the RYR2 gene located in exon 3. A case is presented of a young patient in whom the clinical signs started with recurrent syncope associated with physical or emotional stress. In the subsequent study, catecholaminergic polymorphic ventricular tachycardia was discovered, with areas of non-compacted myocardium and new genetic variant possibly associated with the disease.
La taquicardia ventricular polimórfica catecolaminérgica se produce debido a mutaciones del gen del receptor de la rianodina 2, con la consiguiente alteración del flujo de iones de calcio en el interior de los miocitos ventriculares1. Esta entidad tiene una prevalencia estimada de 1/10.000, afecta a individuos en las primeras décadas de la vida y debuta con palpitaciones, presíncope y síncopes desencadenados por estrés o actividad física2.
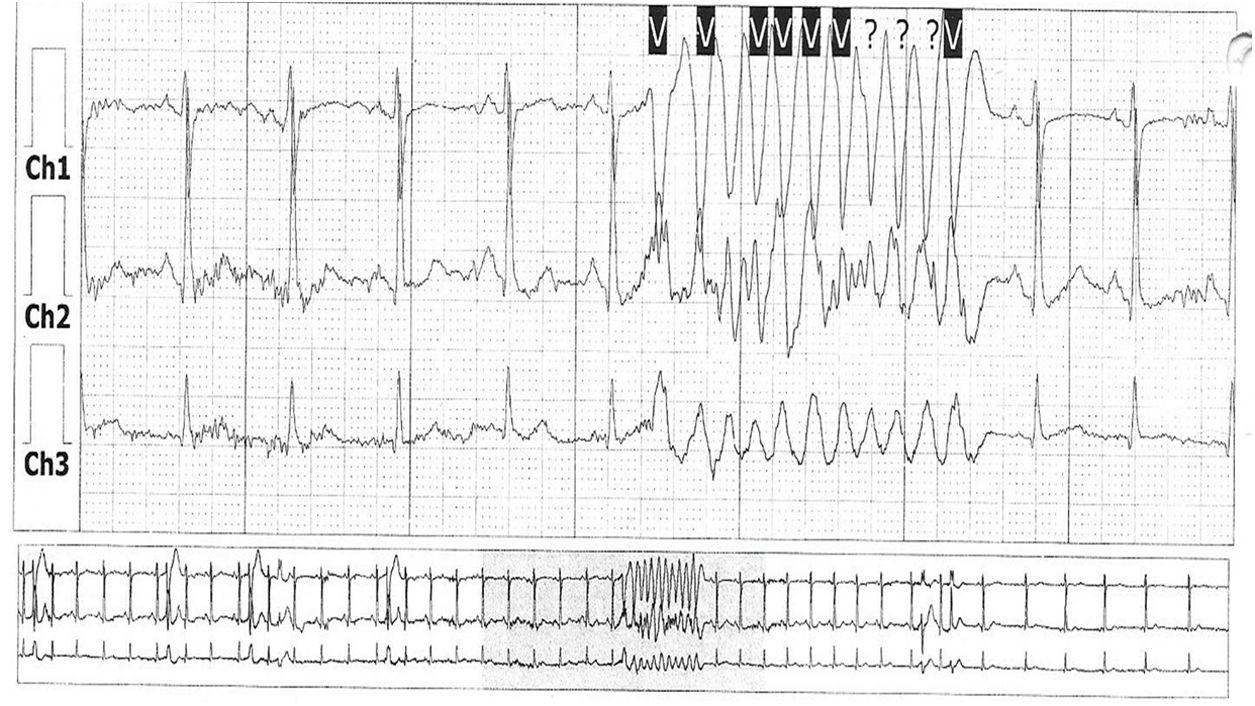
CasoMujer de 31 años, con diagnóstico de síndrome de Gilbert y antecedente de apendicectomía, quien consultó por palpitaciones ocasionales y síncopes de origen indeterminado, recurrentes, desde hacía cinco meses. Normotensa, afebril; auscultación pulmonar rítmica y sin soplos, murmullo vesicular conservado; abdomen no doloroso ni distendido; extremidades inferiores con pulsos periféricos conservados y sin edemas. La analítica de sangre resultó anodina a excepción de una hiperbilirrubinemia indirecta y un hipertiroidismo subclínico. La radiografía de tórax mostró silueta cardíaca normal, sin alteraciones en la trama broncovascular, infiltrados alveolares ni intersticiales y ángulos costodiafragmáticos libres. El electrocardiograma mostró ritmo sinusal a 54 lpm, eje a 60°, PR 124ms y QRS estrecho, sin alteraciones del segmento ST ni de las ondas T. La ecocardiografía transtorácica no mostró valvulopatías, ventrículo izquierdo no dilatado ni hipertrofiado ni se detectaron alteraciones bruscas de la contractilidad segmentaria o derrame pericárdico; la fracción de eyección fue del 62%. Un electrocardiograma Holter de 24 horas mostró extrasístoles ventriculares de alta densidad de varias morfologías; siendo la más frecuente la morfología de bloqueo de rama izquierda del haz de His con eje inferior, con aspecto de taquicardia ventricular helicoidal que parecía originarse por QRS sobre T (fig. 1).

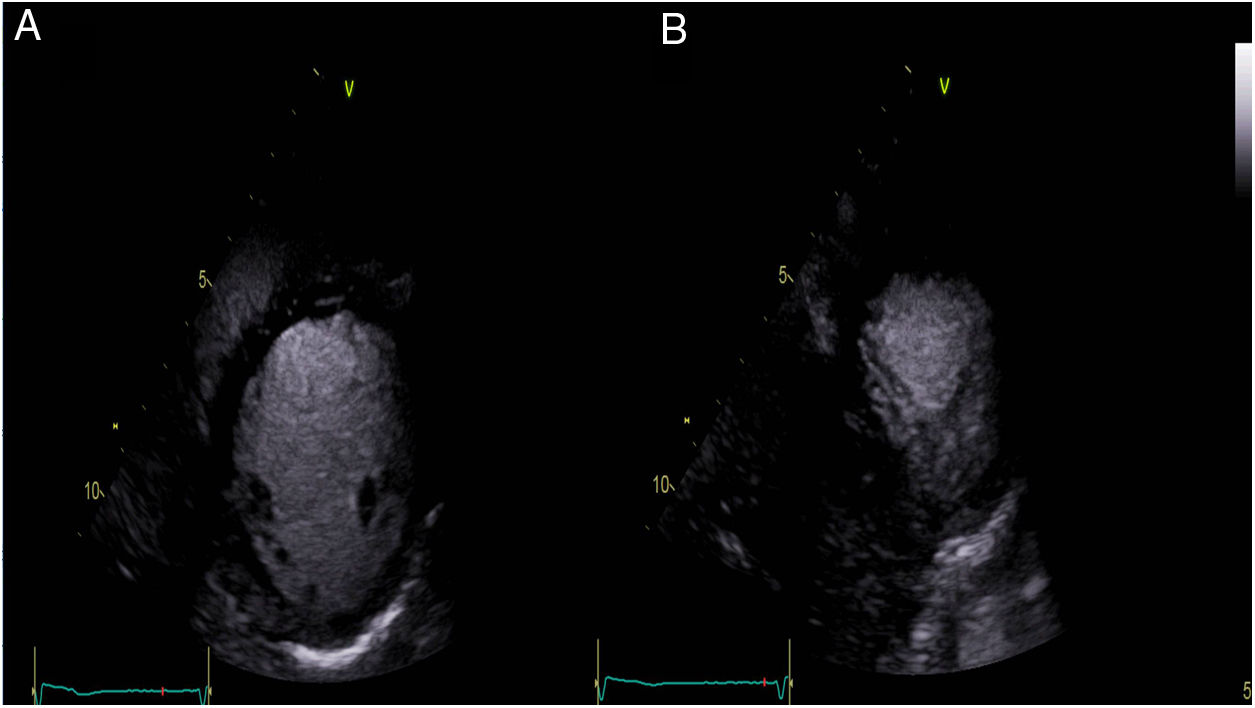
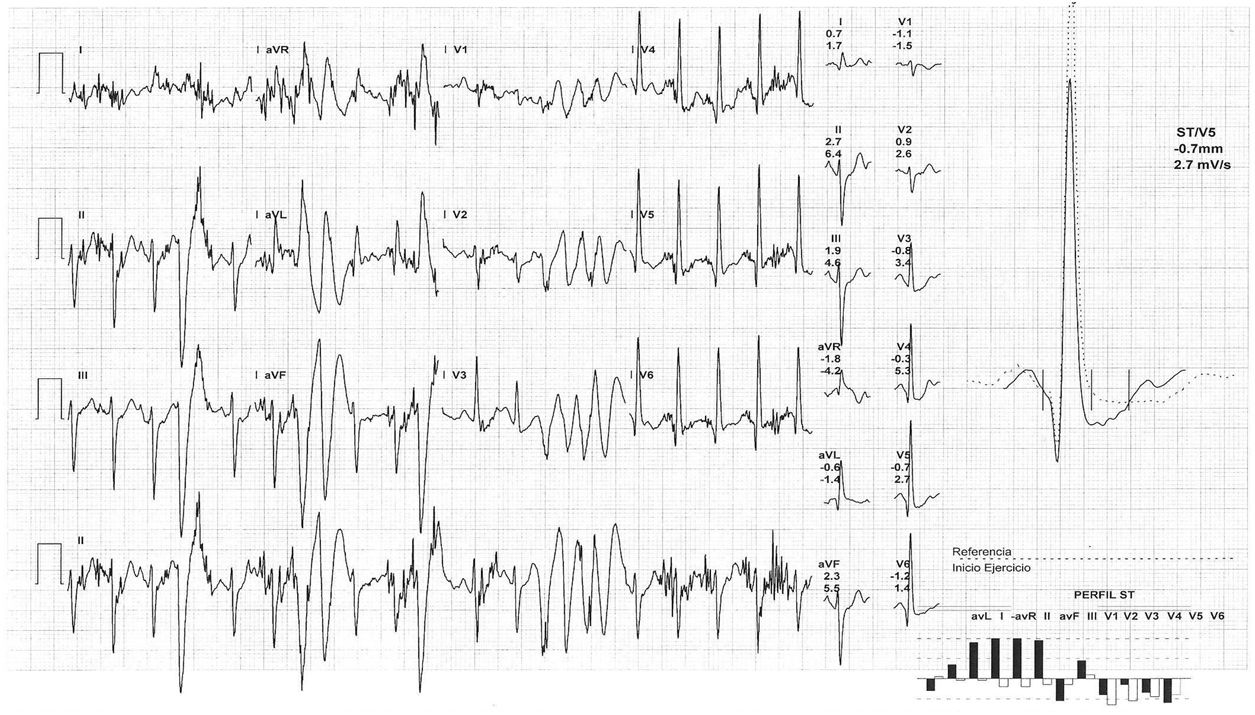
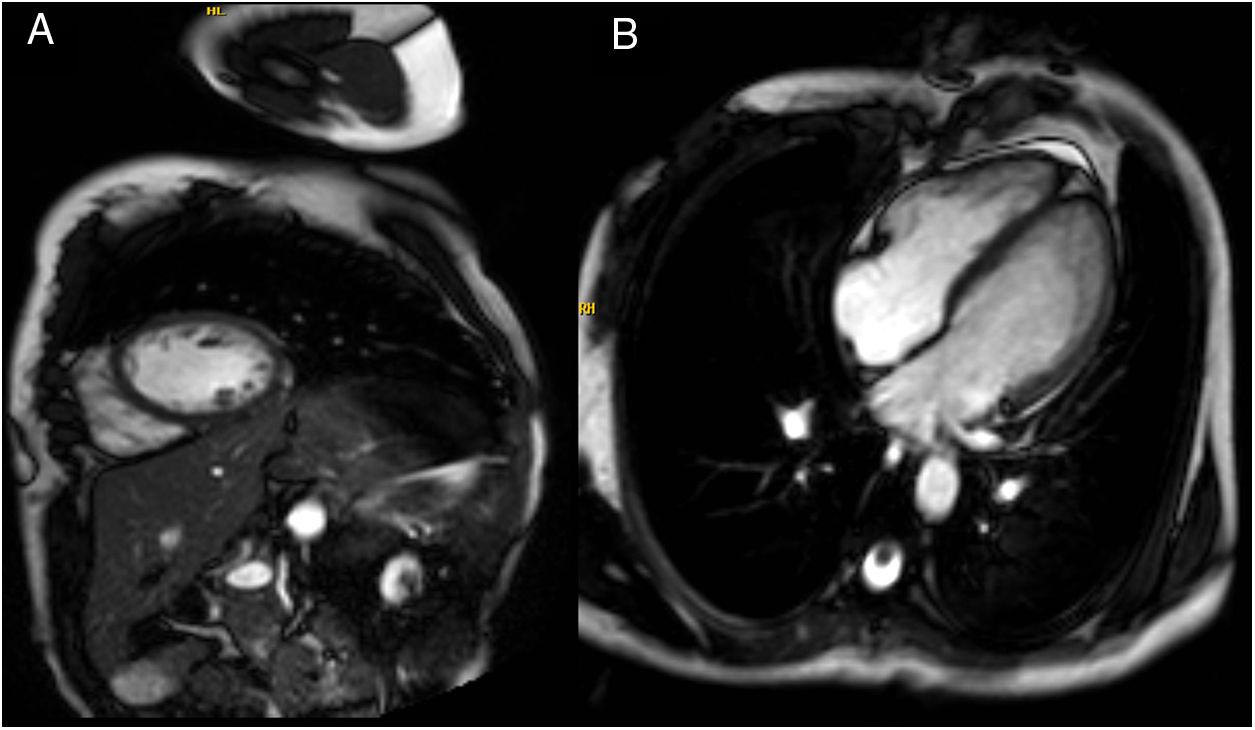
Ante estos hallazgos en el registro de monitorización eletrocardiográfica la paciente fue ingresada en el servicio de cardiología. La determinación de metanefrinas y ácido vanilmandélico en orina de 24 horas fue normal. Durante su estancia permaneció con telemetría; presentó extrasistolia ventricular ocasional, alguna pareja y muy ocasional triplete, sin formas más complejas. Un ecocardiograma transtorácico con contraste objetivó áreas de no compactación en región postero-lateral medio-apical (relación miocardio compactado/ no compactado 1,5 en telesístole y 2,2 en telediástole, límite para criterios de no-compactación) (fig. 2, vídeo 1). Por otra parte, se realizó ergometría simple sin betabloqueadores, que fue concluyente clínicamente negativa y eléctricamente mostró extrasístoles ventriculares de distinta morfología frecuentes desde el segundo estadio de Bruce, que se unen en pares a partir del tercero y salvas de taquicardias ventriculares polimórficas a partir del cuarto (fig. 3). Se inició terapia con propranolol 40mg/8h, con buena tolerancia; la ergometría se repitió pasadas 48 horas y se observó marcada mejoría de las arritmias con extrasistolia y tripletes aislados a alta carga. Así mismo, se practicó cardiorresonancia magnética que confirmó áreas de no compactación en región postero-lateral medio-apical, sin encontrar realce en la secuencia de captación tardía ni hiperintensidad en las secuencias pT2 con supresión grasa y con fracción de eyección del ventrículo izquierdo conservada (68%) (fig. 4).



Previo al alta hospitalaria se implantó un Holter subcutáneo, programando detección de zona de bradicardia a 40 lpm, TV a 171 lpm y TVR a 230 lpm, con el objetivo de valorar la desaparición de las taquicardias bajo el efecto de los betabloqueadores; se solicitó, además, un test genético debido al hallazgo ecocardiográfico de áreas de no compactación ventricular.
El estudio genético reveló que la paciente era portadora de la variante 1:237947553 G/A en heterocigosis del gen RYR2, mutación con cambio de sentido, que condiciona una sustitución de aminoácido glicina por arginina (Gly4181Arg), no descrita previamente como patogénica para la taquicardia ventricular polimórfica catecolaminérgica (resultado no-concluyente). La paciente evolucionó bien y tres meses después se halló asintomática sin datos de recurrencia, a la espera de retirada y revisión del Holter subcutáneo.
DiscusiónLa taquicardia ventricular polimórfica catecolaminérgica es una enfermedad genética, de herencia mendeliana autosómica dominante en su mayoría, producida por mutaciones en el gen RyR2 (1q43), que codifica para la proteína receptora de rianodina 2. Existen más de 70 mutaciones consistentes en sustitución de aminoácidos de la estructura primaria de la proteína, produciendo una alteración estructural del receptor que disminuye la entrada del calcio a través de los canales ICaL de las cisternas terminales de los túbulos T. Ante una alteración del flujo de calcio iónico intracelular sistólico y diastólico, puede producirse una disfunción contráctil del cardiomiocito y generar arritmias ventriculares3. En un porcentaje menor de los casos esta entidad sigue una herencia recesiva causada por mutaciones en otros genes como CASQ2, KCNJ2, Ank2, TRDN o CALM1. El gen CASQ2 codifica para la proteína calsequestrina 2, presente fundamentalmente en las fibras musculares estriadas esqueléticas de contracción lenta. Esta proteína actúa como un ligando del calcio del interior del retículo sarcoplasmático, de tal forma que alteraciones en su estructura o en su función inducen cambios en las concentraciones intracelulares de dicho ion, al igual que ocurría con las mutaciones del gen RYR24.
Los pacientes debutan con palpitaciones, presíncope o síncopes recurrentes, traducción clínica de taquicardias ventriculares inducidas por estrés físico o emocional. La prevalencia estimada es de 1/10.000 y afecta a individuos en las primeras décadas de la vida, si bien se han detectado casos de aparición tardía. El electrocardiograma puede ser anodino y es necesario un estudio de imagen completo para descartar cardiopatía estructural que potencialmente pudiera inducir taquiarritmias. El test de adrenalina (epinefrina) tiene una sensibilidad del 28%, especificidad del 98,2%, valor predictivo positivo del 87,5% y valor predictivo negativo del 75,34%5. El diagnóstico de certeza se establece al hallar taquicardia ventricular polimórfica o bidireccional en ergometría simple6 que generalmente disminuyen en densidad con betabloqueadores, aunque en ocasiones se requiere un estudio genético.
La variante genética de esta paciente se localizaba en el tercer punto caliente (“hotspot”) para mutaciones en el gen RYR2, la cual consistía en un cambio de posición entre un aminoácido glicina por un aminoácido arginina en la posición 4181. El aminoácido Gly4181 se encuentra en la región distal del dominio N-terminal del exón 90 del gen RYR2, donde se encuentra el dominio “I” (aminoácidos 3722-4610), que desempeña un papel importante en la regulación del canal. Aunque en el exón 90 se detecta gran número de variantes patogénicas asociadas a taquicardia ventricular polimórfica catecolaminérgica, esta variante es una de las que aparece en un número más significativo de controles (con una baja pero significativa frecuencia, frecuencia alélica <1/10.000)7.
El manejo clínico consiste en evitar ejercicios físicos intensos y deportes de competición, así como tratamiento con betabloqueadores, especialmente no-selectivos, como propranolol y nadolol8; se pueden añadir calcio-antagonistas no dihidropiridínicos como verapamilo9 o antiarrítmicos tipo Ic como flecainida10 en caso de refractariedad de los síntomas. La disección del ganglio cérvico-torácico (ganglio estrellado) por vía supraclavicular o toracoscópica parece disminuir la tasa de arritmias ventriculares malignas en pacientes con sintomatología refractaria a betabloqueadores y antiarrítmicos, si bien su eficacia no supera el 60% a los dos años11. El implante de un desfibrilador automático debe plantearse en pacientes con síncopes cardiogénicos, taquicardia ventricular bidireccional pese a tratamiento médico óptimo, y en los casos de parada cardiorrespiratoria recuperada12. El tiempo de detección debe ser el máximo posible, con miras a evitar descargas inadecuadas que induzcan liberación de catecolaminas.
El diagnóstico en la infancia y la ausencia de betabloqueadores son factores independientes de síncope, taquicardia ventricular y muerte súbita a largo plazo13. La mayoría de los familiares portadores de mutaciones del gen RYR2 no desarrollan eventos arrítmicos a largo plazo, si bien la localización de la mutación en el dominio C-terminal del gen constituye un factor de riesgo de que éstos se produzcan14. La presencia de taquicardias ventriculares inducidas por ejercicio puede asociarse, en ocasiones, a áreas de no compactación del ventrículo izquierdo, como ocurría en este caso15, incluso la no compactación del ventrículo izquierdo se asocia con mutaciones del gen RYR2. No obstante, el claro desencadenamiento de la taquicardia polimórfica con la ergometría simple, hizo que nos planteásemos la taquicardia ventricular polimórfica catecolaminérgica como opción más probable. En este caso sería interesante evaluar el fenotipo de la taquicardia ventricular polimórfica catecolaminérgica junto con test genético en familiares de primer y segundo grado de la paciente, para ayudar a determinar la patogenicidad de esta variante.
ConclusionesLa presencia de una taquicardia ventricular polimórfica inducida ante situaciones de estrés físico o emocional y desencadenada por una prueba de esfuerzo, como sucedió con esta paciente, establece el diagnóstico de certeza de la taquicardia ventricular polimórfica catecolaminérgica. Tal y como se ha visto previamente, existe asociación en la literatura científica entre esta entidad, áreas de no compactación ventricular y miocardiopatía no-compactada, si bien, en este caso los criterios diagnósticos para esta entidad no llegaban a cumplirse. El manejo clínico empleado consistió en evitar ejercicios físicos intensos y tratamiento con propranolol, con el cual se obtuvo buena respuesta. No fue necesario añadir calcio-antagonistas no dihidropiridínicos ni antiarrítmicos tipo Ic ni proponer el implante de un desfibrilador, debido a la ausencia de refractariedad de los síntomas. La variante genética descrita en la paciente se localizaba en un área del gen RYR2, muy frecuentemente relacionada con mutaciones que inducen taquicardia ventricular polimórfica catecolaminérgica. Por tanto, es posible plantear la relación de esta variante con este síndrome e incluso con la presencia de áreas de ventrículo izquierdo no compactado, a la espera de nuevos reportes en la literatura que la relacionen con alguna de estas patologías.
FinanciaciónLos autores declaran no haber recibido financiación de ninguna entidad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.