




Metachromatic leukodystrophy (MLD) is a rare demyelinating disease (prevalence 1:40,000), also called arylsulfatase A deficiency (ARS-A), which may present with neurological and psychiatric symptoms. Clinical assessment may be difficult, due to unspecific signs and symptoms. A case is presented of a 16 year-old female patient seen in psychiatry due to behavioural changes, psychosis, and with impaired overall performance. She was initially diagnosed with schizophrenia, but the Nuclear Magnetic Resonance (NMR) scan and laboratory tests lead to the diagnosis of MLD.
La leucodistofia metacromática (LDM) es una enfermedad desmielinizante rara (prevalencia, 1:40.000), también llamada deficiencia de arilsulfatasa A (ARS-A), que puede presentarse con síntomas neurológicos y psiquiátricos y cuyo diagnóstico puede plantear dificultades para el clínico, dado lo inespecífico de los signos y síntomas. Se presenta el caso de una paciente de 16 años atendida por psiquiatría por cambios conductuales, psicosis y deterioro general del funcionamiento. Inicialmente diagnosticada como esquizofrenia, se documentaron por resonancia magnética y pruebas de laboratorio en la evolución cambios que llevaron al diagnóstico de leucodistrofia metacromática.
Metachromatic leukodystrophy (MLD) is a demyelinating genetic disorder that can present clinically with neuropsychiatric symptoms such as psychosis, similar to those of patients with schizophrenia.1,2 The late onset of MLD has been suggested as a model of schizophrenia, because both disorders are characterised by signs of generalised anatomical disconnection and secondary functional impairment that indicate an extensive lesion in the frontal-subcortical circuits. These signs are manifested in symptoms affecting mood, motivation, judgement and planning and changes in behaviour,1 suggestive of the recently revived concept of diaschisis, coined by Monakow (1914), in which neurophysiological disorders distant from the focal brain lesion are observed.3
Currently, most patients do not undergo arylsulfatase A gene sequencing, approximately 50% of the alleles have not been identified, and it is not possible to predict clinical outcome based solely on mutation analysis.4
Case reportThe patient first developed behavioural changes at the age of 16, characterised by marked isolation, poor performance and behaviour problems at school, and altered sleep pattern, with psychotic symptoms: soliloquy, hallucinations, disorganised behaviour (eating unpeeled bananas) and general deterioration in functioning, without recovery. We initially requested simple cranial computerised tomography (CT), electroencephalogram, complete blood count, and thyroid and metabolic profile, which were reported as normal. Paranoid schizophrenia was diagnosed. She had several admissions to a mental health unit and had to be institutionalised in a long-term care home as a result of psychotic symptoms and severe episodes of aggression towards others.
Poor pharmacological response with persistent symptoms led to a magnetic resonance imaging (MRI) scan being requested, which showed small nonspecific focal lesions in the white matter of both cerebral hemispheres, and the differential diagnosis included the possibility of ischaemic lesions in small vascular territories. Cerebral angiogram was normal, while the metabolic profile and leptospira, cytomegalovirus and Epstein–Barr virus antibodies were all negative.
The patient was treated with haloperidol and developed extrapyramidal side effects. Subsequently, with olanzapine, she had marked weight gain and with risperidone, amenorrhoea. She was then treated with aripiprazole in progressively higher doses up to 60mg, with which her behavioural and psychotic symptoms were finally stabilised.
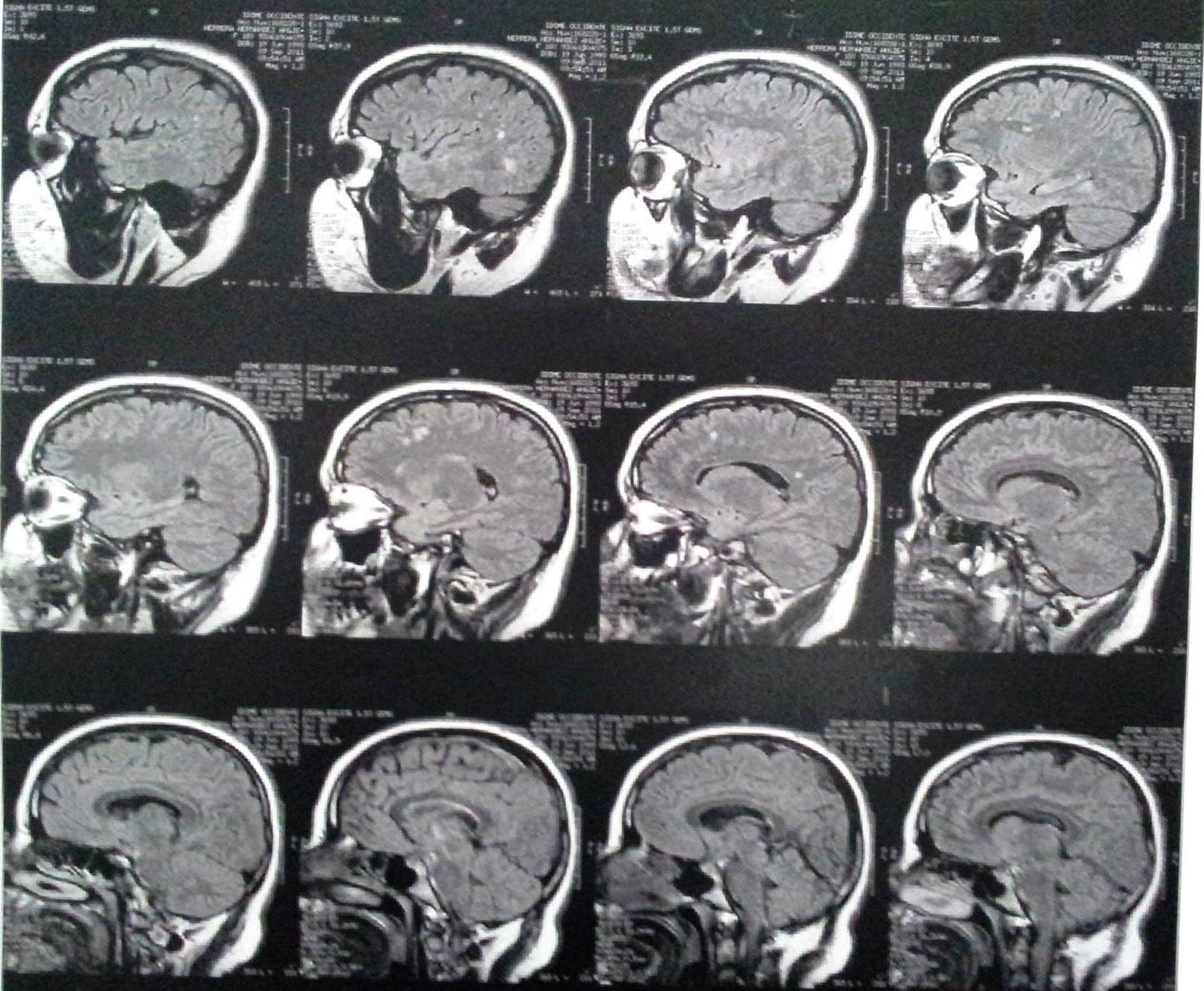
Three years after the first consultation, bilateral T2 hyperintense focal lesions and bilateral FLAIR frontal/parietal subcortical lesions, nonspecific in character, most likely as sequelae, were observed (Fig. 1).

Blood count and thyrotropin, VDRL, renal function and folic acid determinations were normal, while antinuclear and anti-cardiolipin antibodies and the complete phospholipid profile were negative. Rheumatology ruled out autoimmune disease as a cause of the patient's behavioural disorder.
Genetic assessment revealed neurodegenerative disease and white matter disorder. Elevated lactic acid was noted, and metabolic screening for white matter disease was requested. The results showed a significant decrease in arylsulfatase A (ARSA) in leukocytes which, together with the clinical picture and the brain MRI, led to the diagnosis of MLD.
DefinitionMLD is a lysosomal disease of the sphingolipidoses group, caused by a deficiency of ARSA, an enzyme related to the metabolism of sulfates, which is abundant in myelin (Table 1).3 The low concentration of this enzyme affects the metabolism of the cerebroside sulphate and causes intralysosomal storage of sulfatide, also known as 3′O-sulfogalactosylceramide, in the white matter of the central nervous system (oligodendrocytes and glia) and peripheral nervous system, especially in the myelin sheaths that surround the nerve cells and other tissues of the body, such as lactosyl sulfatide of the kidneys, bladder and gallbladder.1,5,6 The accumulation of this material alters the formation and destroys the myelin by an unknown pathophysiological mechanism.7
Classification of gross motor function and expressive language functioning in metachromatic leukodystrophy.15
| Gross motor function (GMFC-MLD) | |
| M0 | Walking without support, with quality and performance normal for age |
| M1 | Walking without support but with reduced quality of performance, i.e., instability when standing or walking |
| M2 | Walking with support; walking without support not possible (fewer than five steps) |
| M3 | Sitting without support but locomotion, such as crawling or rolling, walking with or without support not possible |
| M4 | Sitting with support but locomotion or sitting without support not possible; but locomotion such as crawling or rolling is present |
| M5 | No locomotion nor sitting without support, but head control is possible |
| M6 | Loss of any locomotion as well as loss of any head and trunk control |
| Expressive language functioning (ELFC-MLD) | |
| E0 | Communicates in complete sentences at a quality and performance normal for age |
| E1 | Communicates in complete sentences with reduced quality and performance for age |
| E2 | Cannot communicate in complete sentences, but able to use 2-word phrases |
| E3 | Cannot communicate in 2-word phrases, but able to use single, meaningful words/ideas |
| E4 | Complete loss of expressive language |
MLD is caused by an autosomal recessive mutation of chromosome 22q which results in an ARSA deficiency. A total of 189 mutations have been identified in the ARSA gene,8 including substitution at amino acid 31, one substitution at position, three deletions, three donor site mutations and three donor receptor-binding site mutations.1
Salmon et al. reported the case of a 30-year-old woman with a diagnosis of enzymatically confirmed MLD, with cognitive changes in which bilateral hypometabolism was found in the thalamus, medial frontal cortex, frontal pole and occipital cortex, in contrast to the changes shown in Alzheimer's disease: hypoperfusion in the dorsolateral prefrontal cortex and temporal-frontal lobe.1
Tamagakien reported similar changes in a patient with behavioural changes.1
DiagnosisMLD is suspected when metachromatic granules are found in biopsy of the conjunctiva or sural nerve.1 Diagnosis is confirmed when low ARSA activity is documented in leukocytes or fibroblast culture.1,9
In ARSA pseudo-deficiency, there is a partial deficit that does not cause clinical disorders, and this can complicate the diagnosis and identification of patients with MLD. This condition can also be found in healthy subjects.4
The diagnosis is based on analysis of mutations, biochemistry tests and clinical assessments.4
The tests that can be used for the diagnosis of MLD include the following10:
- •
Blood or skin tests for low ARSA activity.
- •
Brain MRI.
- •
Lumbar puncture to test for high protein concentrations.
- •
Urinalysis for high sulfatide concentrations.
- •
Nerve conduction velocity studies.
Sulfatide in nerve and cerebrospinal fluid and lyso-sulfatide accumulation are markers of severity in peripheral nerve only; they do not reflect the extent of the damage in the central nervous system.11
Prenatal diagnosis and screening of carrier statusBefore birth, ARSA activity can be determined by cell culture of amniotic fluid or chorionic villi. Prenatal diagnosis is indicated for couples who have a history of an affected child. Carrier status can be determined by measuring ARSA activity, although there are values that can be found in the healthy population. It is essential to differentiate the MLD carrier from the benign condition; the most effective way to do this is to test for mutations.4
Differential diagnosisThe radiological changes in MLD can be distinguished from microangiopathy related to ageing or multiple sclerosis, because the lesions in the white matter tend to be symmetrical and confluent.1
It is not uncommon for some patients to be diagnosed with frontotemporal dementia due to alterations in attention, language, information processing and executive functions.2 The behavioural changes associated with subtle symptoms of memory deficit and motor alterations can suggest the diagnosis of early-onset dementia.1
In adolescents, MLD can resemble a psychosis because the disease alters the critical process of myelination, especially in the frontotemporal anatomical connections. This triggers alterations in the structure and function of the central nervous system, and can lead to symptoms similar to those of schizophrenia.2
Imaging findingsMRI findings include hyperintense areas of diffuse, bilateral, and often symmetrical demyelination in the periventricular white matter and cerebellum that may converge with disease progression, with frontal predominance in the late stages (juvenile forms and in adults).2,7,12
On CT, hyperdensities are observed in white matter, particularly in the frontal and parietal regions. As it progresses, cortical and subcortical atrophy is observed, with ventricular dilatation. Areas of hypodensity reflect loss of myelination and cerebroside accumulation.1 A “tigroid” or “leopard skin” appearance has been described, with hypointense bands (normal white matter) within T2 hyperintense areas of abnormal white matter (areas of demyelination).7
There is evidence of significant loss of grey matter volume from early stages of MLD. In adults, there is a more pronounced general cortical atrophy of both grey and white matter and a more pronounced cortical reduction in cingulate gyrus and frontal lobes.13
MRI is more sensitive than CT in the detection of lesions in the white matter and it is useful for visualising early-onset lesions in regions of the posterior fossa (brain stem or cerebellum); it also shows the severity and extent of the disease.1
There is evidence of significant loss of cortical grey matter in the early stages of the disease. In the adult forms, there is greater general cortical atrophy, more pronounced in cingulate gyrus and frontal lobes.13
MRI spectroscopy usually shows a decrease in N-acetylaspartate and myo-inositol and, occasionally, an increase in lactate.7
EpidemiologyMLD occurs with an estimated frequency of 1:40,000,1,5 although it is estimated that up to 16% of the general population may have an ARSA deficiency.1
Presentation and clinical courseBecause it is a rare, heterogeneous disease with diverse clinical presentation and lack of clinical, neurophysiological and neuroradiological documentation, little is known about factors related to age of onset and the course of the disease, making it difficult to predict how each particular case will progress.4
The different ways in which the disease progresses are related to the type of mutation.5 Like all metabolic diseases, MLD can cause serious damage to the central nervous system, especially in the neurons which are sensitive to metabolic changes. This may manifest itself in impaired psychomotor development, seizures and coma when the lesion is severe, and with subtle symptoms such as cognitive and behavioural alterations when the lesion is mild.2 There is a clinical classification of motor and language disorders, referring to the severity of symptoms (Table 1).
Depending on the age of onset, they are classified into late-infantile, juvenile, late-juvenile and adult subtypes.1,5,6,9
Late-infantile subtypeThe late-infantile subtype, described by Greenfield14 in 1933, is the most prevalent. Onset is around the first and second year of life and there are several stages. In the first stage, it begins with motor impairment such as ataxic gait or loss of walking ability, hypotonia and abnormal deep tendon reflexes. This stage has an average duration of 16 months. In the second stage, the motor impairment becomes more severe and is accompanied by mental impairment. This stage lasts from three to six months. During the third stage, patients develop tetraplegia, bulbar paralysis, atrophy of the optic nerve and central nervous system involvement. This stage lasts from three months to three years, followed by a vegetative state that can last for years and usually culminates with death at around the age of four or five. ARSA concentrations are low or non-existent.1,6
Juvenile subtypeIn the juvenile form, the age of onset is usually between 4 and 6 years old. It presents with mental alterations, such as emotional lability, euphoria and behavioural changes, loss of mental functions and language difficulties that affect intellectual development and school performance. In cases with slow progression, motor symptoms predominate; spastic tetraplegia, bulbar symptoms, ataxia, seizures and atrophy of the optic nerve may all occur. Disease duration can vary from 3 to 17 years.1,6 ARSA enzyme activity is limited, but not so limited as in the infantile form.5
Late-juvenile subtypeIn the late-juvenile subtype, the disease usually begins between the ages of 6 and 16 years and progresses slowly. It usually presents with behavioural problems, slowly evolving mental disorders and seizures or motor problems. ARSA shows residual activity.1,5
Adult subtypeIn the adult subtype the symptoms appear after the age of 16 years. It may present in two different ways: one with motor predominance (pyramidal cerebellar syndrome); and one with predominance of psychiatric symptoms. This late form is strongly associated with I179S mutation.1,2
In more than half of patients the age of onset of the disease is between 10 and 30 years, with symptoms similar to psychosis, including auditory hallucinations, delusions, altered thought processes and catatonia.2
The disease progresses with neurological symptoms such as seizures, chorea and dystonia that occur after the onset of psychiatric symptoms.2
TreatmentThere is currently no specific treatment for this disease, except bone marrow transplantation in selected cases.
The indicated treatment is symptomatic and supportive; physiotherapy, respiratory therapy, nutrition, nursing care, adaptation of the environment and maintaining the support network and usual activity are indicated.4 The use of anti-epileptic and antispasmodic drugs has also been indicated.4
Gene therapy has been shown to be more effective than traditional healthy donor transplantation to correct the disease.4
Haematopoietic stem cell transplantation (HSCT) is a viable option, although with limitations, which may benefit late-onset phenotypes in the early, pre-symptomatic phase.15
DiscussionCase reports, case series and cohorts have already been published.14,16–18 The case presented here is a late-juvenile MLD with the nonspecific onset of the symptoms which was initially suggestive of psychotic disorder. Initial tests were negative, including diagnostic imaging. Inadequate symptom control and deteriorating condition led to the need for permanent institutional care. Later, the appearance of motor signs led to a neurodegenerative disease being suspected, and the diagnosis was made with the participation of psychiatry, neurology and genetics departments. The initial picture, given the patient's age and symptoms, suggested the onset of a schizophrenia-like psychotic illness, although its progression and lack of therapeutic response led to the diagnosis being reconsidered.
Although cases of MLD have been reported in relatives,18 most cases, such as the one presented here, correspond to only one affected person in the genogram. The initially negative findings in diagnostic imaging and the subsequent presence of lesions on the MRI showed the clinical progression.
Psychiatrists should be aware of their role in early diagnosis of MLD, since initial symptoms may lead to psychiatric consultation. Although its prevalence is low, the onset may be similar to that of schizophrenia and other psychotic disorders. Adequate follow-up should improve the chances of earlier diagnosis and management.
Although there is no evidence of any effective treatment for the condition, and symptomatic treatment and supportive measures are assumed to be key, side effects of medication should be avoided as much as possible.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Please cite this article as: Espejo LM, de la Espriella R, Hernández JF. Leucodistrofia metacromática. Presentación de caso. Rev Colomb Psiquiat. 2017;46:44–49.
