



Schoenlein-Henoch purpura is a systemic small vessel vasculitis mediated by IgA-1 deposition in organs such as the skin, kidney, and gastrointestinal tract; it has been mainly described in children where it has a favourable prognosis. Although much rarer in adulthood it is associated with an increased risk of severe kidney involvement, gastrointestinal complications, and prolonged hospital stay. The therapeutic options are wide and vary according to the degree of involvement of the patient and the organ mainly affected.
La púrpura de Schoenlein-Henoch es una vasculitis sistémica de pequeño vaso mediada por depósito de IgA en órganos como la piel, el riñón y el tracto gastrointestinal. Se ha descrito principalmente en niños, grupo de población en el que tiene un pronóstico favorable. Si bien en la edad adulta es mucho menos frecuente, se asocia con un mayor riesgo de compromiso renal severo, complicaciones gastrointestinales y estancia hospitalaria prolongada. Las opciones terapéuticas son amplias y varían según el grado de compromiso del paciente y el órgano más afectado.
Schönlein-Henoch purpura is a small-vessel vasculitis, mediated by deposits of immunoglobulin A (IgA), with multisystem involvement, which mainly compromises the skin, the gastrointestinal system, the kidneys and the joints.1 Cutaneous involvement, which is frequent and is usually the first manifestation of the disease, is characterized by the presence of palpable purpura with histopathological representation of leukocytoclastic vasculitis.2
Although it is a frequent condition in children, its development in adulthood is usually associated with higher rates of kidney injury, gastrointestinal complications, recurrence of lesions after the first episode, and length of hospital stay.3
A series of four cases of Schönlein-Henoch purpura in adult patients with varying degrees of systemic involvement, managed in a high complexity referral center, is described below.
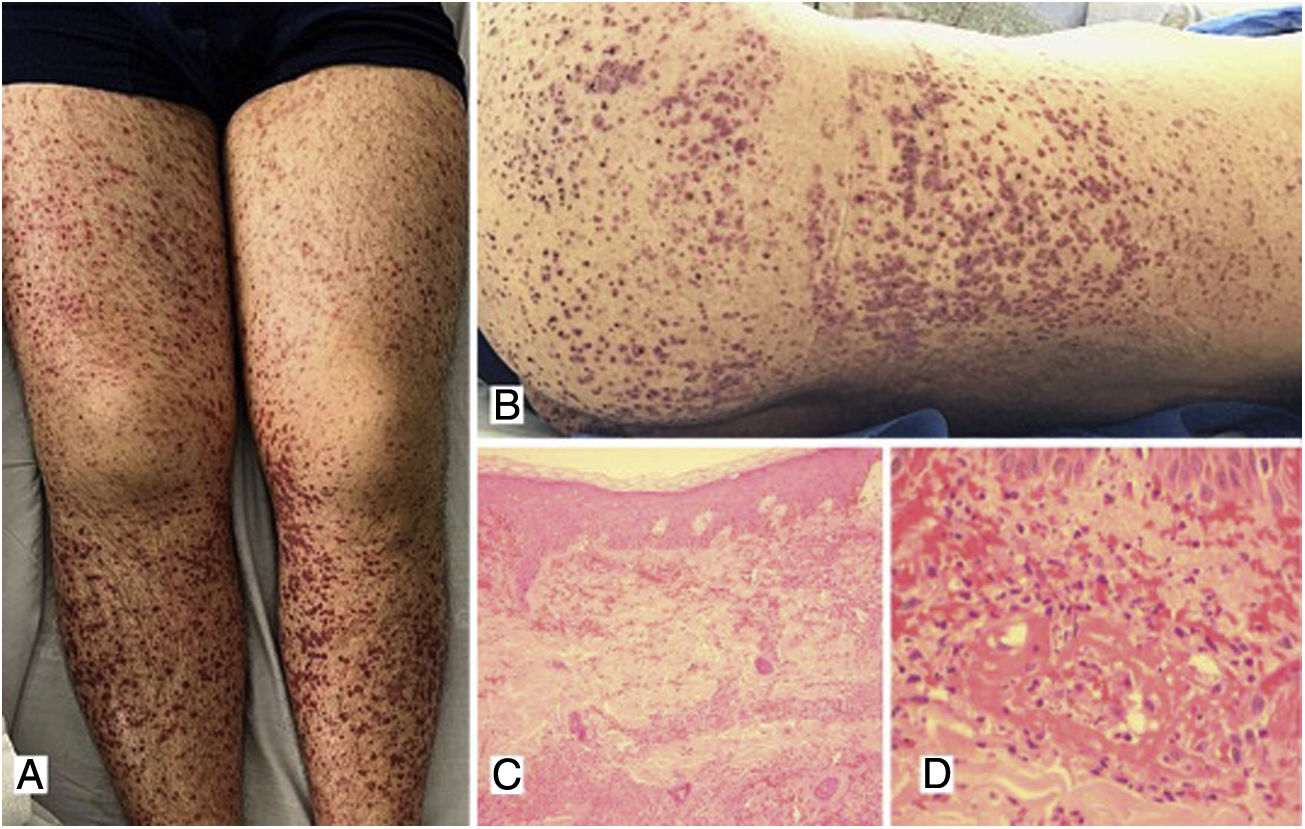
Case 1A 39-year-old male patient with a history of arterial hypertension (AHT) since he was 18 years old, being managed with enalapril, with no prior study of secondary causes. The patient consulted due to a clinical picture of three weeks of evolution of abdominal pain associated with liquid stools, without mucus or blood, with subsequent appearance of violaceous lesions in the lower limbs which later spread to the abdomen and thorax, he also presented with edema of the lower limbs and polyarthralgia of small joints in the hands. The physical examination revealed palpable purpura (Fig. 1) and arthritis with joint effusion. The paraclinical tests showed leukocytosis with neutrophilia, normal liver and kidney function, and a negative infectious profile. A skin biopsy was performed, which was compatible with leukocytoclastic vasculitis, and direct immunofluorescence showed deposits of IgA. The patient evolved satisfactorily after starting oral steroids, with no evidence of renal involvement during his hospital stay.
 Multiple purpuric macules and papules that do not disappear on digital pressure in the lower limbs, buttocks and posterior trunk, some with hematic crust on their surface. C) Skin biopsy with evidence of leukocytoclastic vasculitis and fibrinoid necrosis in superficial and deep dermal vessels. H&E 10×. D) Extravasation of erythrocytes with fibrinoid necrosis of small dermal vessels. H&E 40×.")
A, B) Multiple purpuric macules and papules that do not disappear on digital pressure in the lower limbs, buttocks and posterior trunk, some with hematic crust on their surface. C) Skin biopsy with evidence of leukocytoclastic vasculitis and fibrinoid necrosis in superficial and deep dermal vessels. H&E 10×. D) Extravasation of erythrocytes with fibrinoid necrosis of small dermal vessels. H&E 40×.
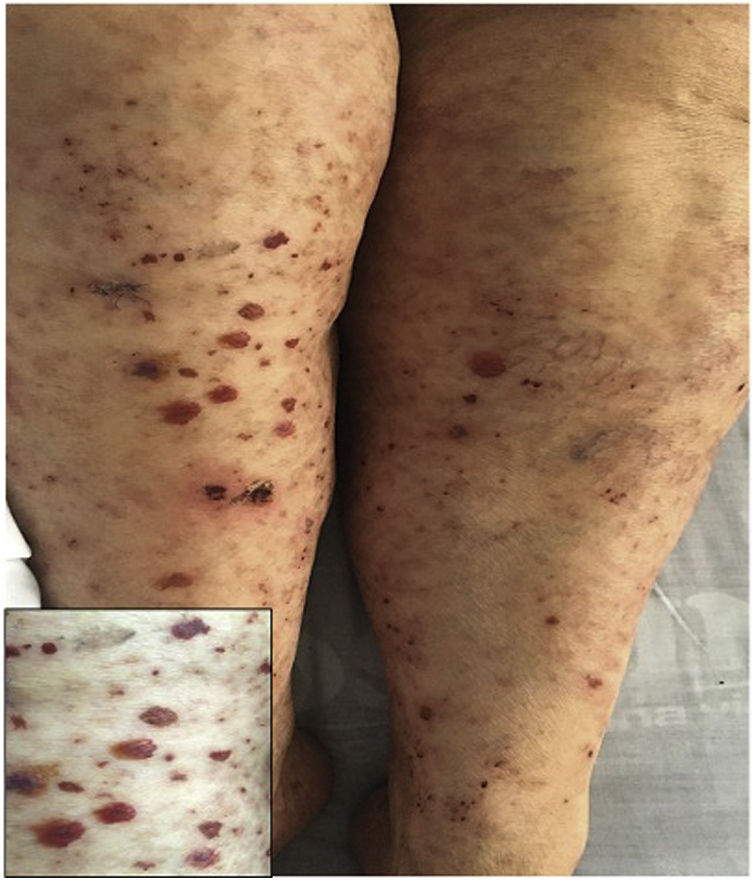
A previously healthy 74-year-old female patient, with a clinical picture of one day of evolution of appearance of purpuric lesions in the lower limbs that ascendingly extended to the infraumbilical region and the upper limbs; associated with this, she presented foamy urine, choluria and arthralgia, predominantly in the knees. On physical examination, she had palpable purpura in the lower limbs (Fig. 2). The paraclinical tests showed negative infectious and autoimmune profiles, preserved renal function, but active sediment in the urinalysis and proteinuria in the nephrotic range.

A renal biopsy was performed with evidence of IgA nephritis/vasculitis-Schönlein-Henoch purpura, without findings of superimposed podocytopathy. The skin biopsy reported leukocytoclastic vasculitis. With these findings, it was decided to start methylprednisolone in bolus and subsequently oral steroid, with satisfactory clinical evolution.
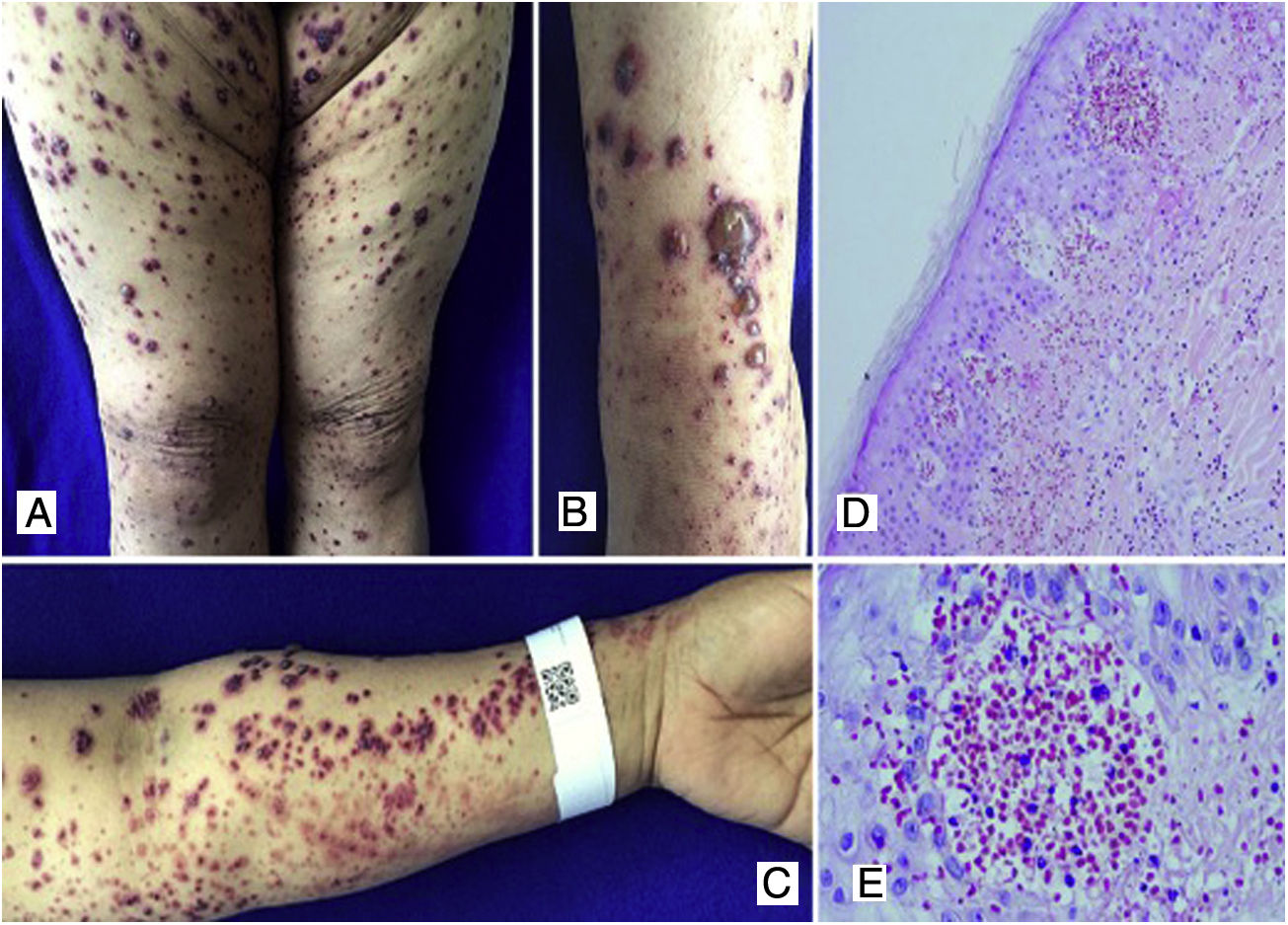
Case 3A 50-year-old female patient who consulted for skin lesions on the legs, arms, abdomen, and face of a week of evolution, associated with intense epigastric pain. In the previous days, she presented arthralgia and swelling in the wrists, elbows and knees. On admission, she presented palpable purpura on the extremities, abdomen, buttocks, and face (Fig. 3). The laboratories reported an elevated erythrocyte sedimentation rate (ESR) and urinary sediment without activity. The patient was taken to an upper digestive endoscopy which reported ischemic duodenitis. A skin biopsy was taken with a report of leukocytoclastic vasculitis and direct immunofluorescence positive for IgA. The immunological profile was negative.
 Multiple purpuric macular plaques and blisters with hemorrhagic content located on the lower and upper limbs, scarce pustules. D) Skin biopsy with perivascular polymorphonuclear inflammatory infiltrate, intraepidermal erythrocyte extravasation and scarce fibrinoid necrosis of superficial dermal vessels. H&E 10×. E) Extravasation of erythrocytes. H&E 40×.")
A, B, C) Multiple purpuric macular plaques and blisters with hemorrhagic content located on the lower and upper limbs, scarce pustules. D) Skin biopsy with perivascular polymorphonuclear inflammatory infiltrate, intraepidermal erythrocyte extravasation and scarce fibrinoid necrosis of superficial dermal vessels. H&E 10×. E) Extravasation of erythrocytes. H&E 40×.
Schönlein-Henoch purpura with extensive skin, joint, and gastrointestinal involvement was diagnosed; the patient was treated with systemic corticosteroids, initially with pulses of methylprednisolone, followed by prednisolone at a dose of 1mg/kg/day, with good clinical response and no need for other therapies.
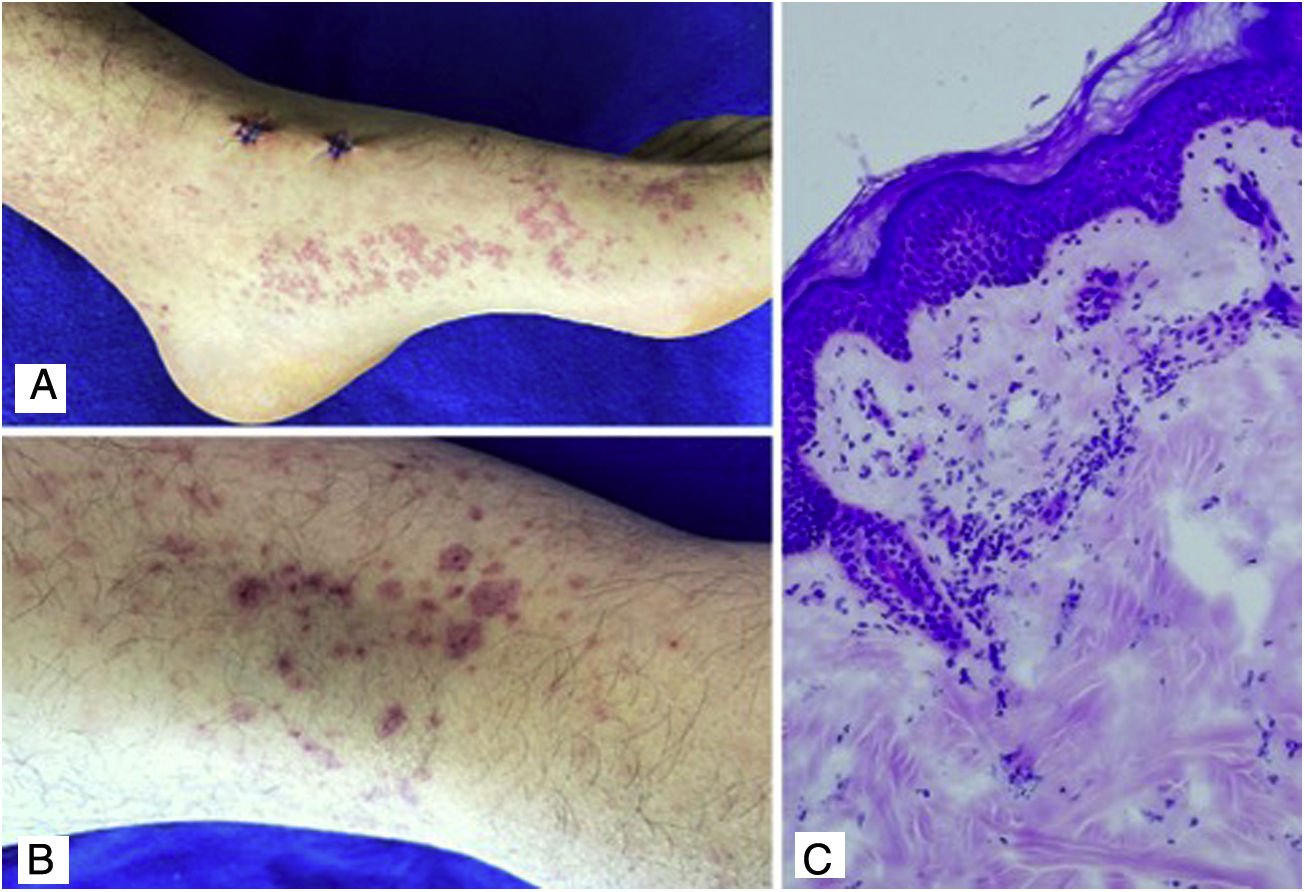
Case 4A 24-year-old male patient who consulted for violaceous lesions in the lower limbs, buttocks, genital region, and upper limbs of two days of evolution, associated with colicky abdominal pain, foamy urine, and joint pain in the hands and knees. On physical examination, lesions compatible with palpable purpura were found (Fig. 4), a tender abdomen on diffuse palpation, without signs of peritoneal irritation. The laboratory tests reported nephrotic proteinuria, nitrogen compounds within the normal range, negative hepatotropic viruses, and a negative autoimmune profile. The skin biopsy reported leukocytoclastic vasculitis, direct immunofluorescence without IgA deposits; however, the renal biopsy reported extracapillary proliferation of less than 25%, moderate mesangial proliferation, endocapillary proliferation and segmental sclerosis, and immunofluorescence with IgA deposits. The diagnosis of systemic IgA vasculitis was confirmed and, taking into account the results of the renal biopsy, management with systemic corticosteroids at a dose of 1mg/kg/day was decided, without the association of another immunomodulator, with an adequate therapeutic response.
Discussion Purpuric macules and plaques with hematic crust that do not disappear on digital pressure located in the lower limbs. C) Skin biopsy with polymorphonuclear perivascular inflammatory infiltrate. H&E 40×.")
Schönlein-Henoch purpura is a small-vessel systemic vasculitis caused by deposition of immune complexes and immunoglobulin A-1 in the walls of arterioles, venules, and capillaries; it is characterized by its multiorgan involvement, with a combination of skin, joint, gastrointestinal and renal commitment, the last two being the main causes of morbidity and mortality in adults.4
This disease has an annual incidence that varies between 3 and 26 cases per 100,000 people and is more frequent in children between four and seven years of age. It is considered the most frequent cutaneous vasculitis in the pediatric age,5 in contrast, in adults it occurs with an approximate annual incidence of 0.1–1.8 cases/100,000 individuals.6
Its exact cause is unknown, it has been proposed a defect in clearance of IgA1 or aberrant glycosylation thereof,7 likewise, it has been raised the participation of certain subtypes of human leukocyte antigens (HLA), such as HLA-DRB1*01, in which certain infectious agents (bacteria, viruses or parasites) can act as triggers, which would explain that up to 40% of patients have a history of upper respiratory infection prior to the development of the vasculitis.4,7 It is suggested that infectious processes trigger the formation of antigen-antibody complexes, which are deposited in the wall of small vessels and thus activate the alternative complement pathway, which generates activation and accumulation of neutrophils without a granulomatous reaction.5
The clinical manifestations are broad and heterogeneous. The disease is characterized by the classic tetrad of palpable nonthrombocytopenic purpura, arthritis or arthralgia, and renal and gastrointestinal involvement.8 Infrequently, other organs and systems such as the lungs, the genitourinary tract, and the central nervous system may be involved.5
Cutaneous involvement is usually the first manifestation of the disease and presents itself as symmetrical palpable purpura, located in areas of pressure and in the lower limbs, which may coalesce to form large plaques, in adults, up to 35% of the cases occur with necrotic or hemorrhagic lesions.3 The skin biopsy evidences leukocytoclastic vasculitis, however, the findings do not differentiate it from other causes of small vessel vasculitis. The deposit of IgA in direct immunofluorescence is highly suggestive, with a specificity that varies between 89 and 100% and with a positive predictive value of 84%; it is recommended to perform it when there is any diagnostic suspicion.
It has been observed that a positive immunofluorescence has an impact on the prognosis of the patients, being associated with more severe and chronic cutaneous manifestations and a higher risk of gastrointestinal and renal compromise.6
The presence of arthralgia is found in two thirds of patients and mainly affects the ankles and the knees, although it may be associated joint edema and effusion.3
Gastrointestinal involvement is secondary to intestinal ischemia and edema and is manifested as colicky abdominal pain in 100% of the cases, nausea and vomiting in 14%, and melena and rectorrhagia in 12.9% of the cases. The main complications include intussusception, intestinal infarction and perforation.7
Renal compromise is evidenced in 50%–85% of the patients, manifested as macroscopic hematuria, which is the earliest and most sensitive sign. It can coexist with proteinuria, macroscopic hematuria, arterial hypertension, and renal failure in up to 30% of cases, the latter being exceptional in children, with reports of less than 2% of cases.4
Other less reported manifestation include myocarditis, orchitis, episcleritis, alveolar hemorrhage, seizures, pancreatitis, parotitis, and myositis.3
The prognosis in adults is unpredictable, it has been shown that up to 13% evolve to severe kidney disease with the need for renal replacement therapy, even in oligosymptomatic patients, and up to 20% of patients have relapses of the disease with reappearance of skin lesions and gastrointestinal or renal manifestations.4,6 Age over eight years, the number of relapses, elevated creatinine at the onset of the clinical picture, proteinuria higher than 1g/day, arterial hypertension, purpura above the waist, persistent skin involvement, increased ESR and increased serum concentration of IgA, with reduced concentration of IgM at the time of diagnosis have been described as conditions predictive of a poor prognosis.5,6
Regarding the classification and diagnostic criteria, the first ones were suggested in 1990 by the American College of Rheumatology (ACR), with a specificity of 87.1% and a sensitivity of 87.9%,9 later, in 1992, they were reviewed in the Chapell Hill consensus,2 and in 2010, the EULAR/PRINTO/PRES (European League against Rheumatism/Pediatric Rheumatology International Trials Organization/Pediatric Rheumatology European Society) criteria were published, with a sensitivity of 100% and a specificity of 87%. These criteria are the most current to date, but is important to clarify that they have only been validated in the pediatric population,1 and are summarized in Table 1.
EULAR/PRINTO/PRES criteria for the diagnosis of Schönlein-Henoch purpura.
| [0,2-3]EULAR/PRINTO/PRES criteria | |||
|---|---|---|---|
| Criterion | Definition | Sensitivity (%) | Specificity (%) |
| Purpura (mandatory criterion) | Purpura (commonly palpable) or petechiae, predominantly in the lower limbs, not associated with thrombocytopenia | 89 | 87.5 |
| - For atypical purpura, a skin biopsy with the presence of IgA deposits is required | |||
| Abdominal pain | Diffuse colicky abdominal pain, of acute onset, documented by medical history or by physical examination. It may be associated with intussusception and gastrointestinal bleeding | 61 | 64 |
| Arthritis or arthralgia | Acute onset arthritis defined as joint effusion or joint pain with limited movement | 78 | 42 |
| Renal involvement | - Proteinuria>0.3g/24h or albumin/Cr ratio>30mmol/mg in isolated urine sample | 33 | 70 |
| - Hematuria, defined as >5 red blood cells per HPF, or 2 or more red blood cell casts | |||
| Histopathology | Leukocytoclastic vasculitis with predominance of deposits of IgA, or proliferative glomerulonephritis with predominance of deposits of IgA | 93 | 89 |
| EULAR/PRINTO/PRES criteria | Skin lesions already described as a mandatory criterion and at least one of the other four criteria already mentioned | 100 | 87 |
Source: Adapted from: Ozen et al.1
The disease tends to be self-limited, many patients resolve symptoms spontaneously and only require symptomatic management for the rash and arthralgia,10 however, it has been reported that the disease is more aggressive in adults, with a higher risk of renal compromise, and it is also difficult to predict the course of the pathology. There is the case of patients with severe disease who remit spontaneously and others who with apparently benign initial courses progress to aggressive kidney disease.6,11
In patients with severe manifestations, the first-line management is systemic corticosteroids at initial doses of 1−2mg/kg/day. Methylprednisolone pulses are indicated in severe and life-threatening situations such as renal involvement with rapidly progressive glomerulonephritis, central nervous system commitment, and risk of gastrointestinal complications such as GI bleeding, mesenteric ischemia, and intussusception.5,12
Management with other immunosuppressive therapies such as cyclophosphamide, cyclosporine, mycophenolate mofetil, and azathioprine has been described in cases refractory to the use of corticosteroids.5Table 2 summarizes the therapeutic options available.
Treatment of IgA vasculitis.
| Treatment of IGA vasculitis | ||
|---|---|---|
| Drug | Indication | Observations |
| Mild rash, arthritis | ||
| Oral corticosteroids | Severe rash, skin edema, abdominal pain, scrotal or testicular involvement | Symptomatic management, shorter duration of symptoms when compared to placebo. They do not prevent systemic compromise |
| IV corticosteroids | Same indications as oral corticosteroids, in patients who do not tolerate the latter | |
| High doses of IV corticosteroids | Nephrotic-range proteinuria | Decreases progression to end-stage renal disease in some case series |
| High doses of IV corticosteroids and immunosuppression | Rapidly progressive glomerulonephritis, diffuse alveolar hemorrhage, CNS involvement | Grade D recommendation |
| Plasmapheresis or IV immunoglobulin | Disease refractory to combined therapy of corticosteroids and immunosuppression, massive gastrointestinal hemorrhage or bleeding of other organs | Grade D recommendation |
Source: Adapted from: Audemard-Verger et al.6
Although there is no solid evidence to date, in certain cases associated with alveolar hemorrhage, severe renal compromise and severe involvement of the central nervous system, the use of plasmapheresis and IV immunoglobulin has been proposed, with contradictory results.6
ConclusionsSchönlein-Henoch purpura is a small-vessel vasculitis that commonly affects the pediatric population and is characterized by the tetrad of palpable purpura, arthritis, gastrointestinal symptoms, and renal involvement. Despite its low frequency in adults, it should be suspected in all patients with these symptoms, being skin involvement generally the first to appear and the most severe, as reported in our patients; the risk of complications and fatal outcomes in this age group makes timely diagnosis and treatment imperative. Systemic steroids continue to be the first line of therapy with an adequate response, as evidenced in the previous cases, in which there was no need to escalate to other immunosuppressive therapies.
Ethical considerationsThe research project was approved by the Research Ethics Committee of the Colombian National University Hospital (Hospital Universitario Nacional de Colombia) (CEI-HUN). The authors of the work declare that they have the signed consent of the patients.
FundingNo funding sources are reported.
Conflict of interestNo conflicts of interest are reported.
