



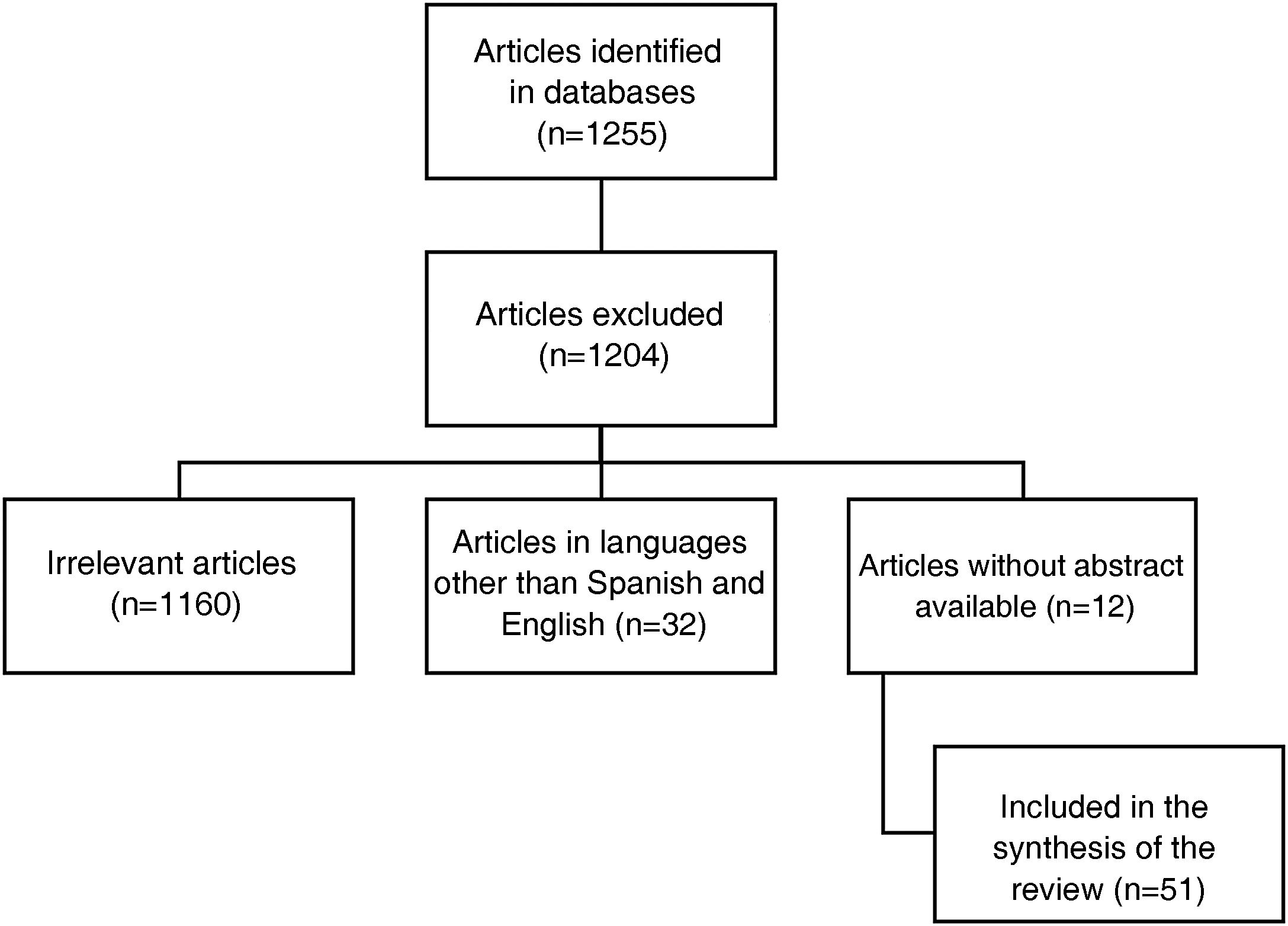
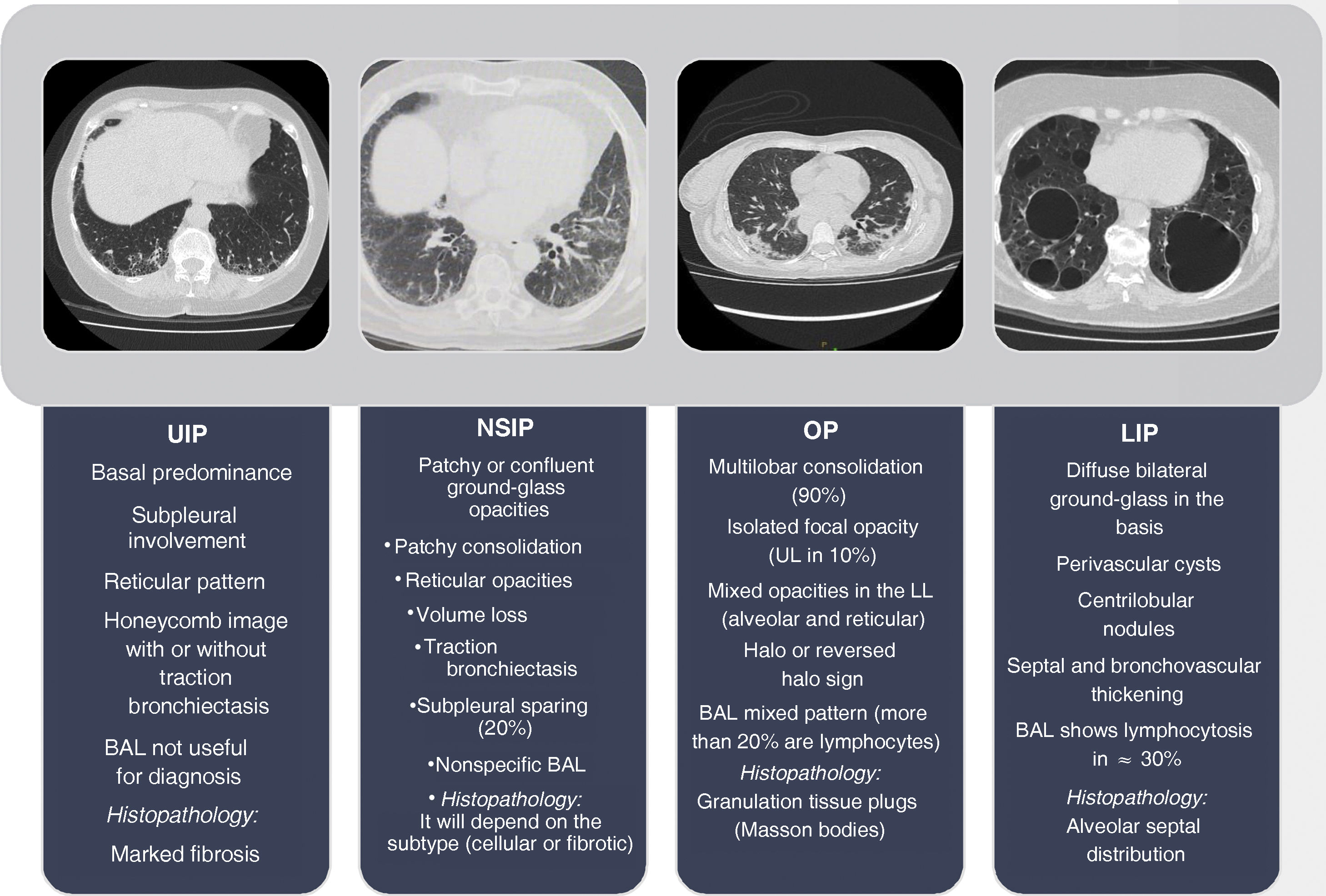



Interstitial lung disease in lupus is an entity that occurs infrequently and tends to progress slowly in most cases. Despite this, the therapeutic approach for moderate to severe cases is largely unknown because most of the evidence comes from case reports, many of which predate the advent of today’s known treatments for lupus. Additionally, little progress has been made in understanding its pathophysiology and current concepts come from other connective tissue diseases such as systemic sclerosis or are grouped within the group of interstitial pneumonias with autoimmune characteristics. This, to an extent, has been an obstacle for research in this field, and to date there is no unified diagnostic and therapeutic approach. Therefore we conducted a state-of-the-art search of the best evidence available to date, in terms of diagnostic methods and emerging therapies, to offer the clinician a practical vision for a comprehensive approach.
La enfermedad pulmonar intersticial en el lupus es una entidad que se presenta con poca frecuencia y en la mayoría de los casos tiende a ser de progresión lenta. A pesar de esto se desconoce en gran medida el enfoque terapéutico de los casos moderados a severos debido a que la mayor parte de la evidencia proviene de reportes de caso y muchos de ellos fueron anteriores al advenimiento de los nuevos tratamientos para el lupus que se conocen hoy en día. Adicionalmente, se ha avanzado poco en entender su fisiopatología, los conceptos actuales provienen de otras enfermedades del tejido conectivo como la esclerosis sistémica que en ocasiones son agrupadas dentro del grupo de neumonías intersticiales con características autoinmunes. Esto de cierta forma ha sido un obstáculo para la investigación en este campo, sin que se haya logrado un enfoque diagnóstico y terapéutico unificado. Por ello, se realiza una búsqueda avanzada con el objetivo de tener la mayor evidencia disponible hasta la fecha en cuanto a los métodos diagnósticos y terapias emergentes, ofreciendo al clínico una visión práctica para lograr su abordaje integral.
