



Juvenile localized scleroderma is a polymorphic disease. It is more prevalent in girls and has a significant morbidity. Extra-cutaneous involvement is common, and polyautoimmunity can reach 7%. The clinical characteristics of this disease in Colombian patients are currently unknown.
ObjectiveTo describe the clinical characteristics, morbidity and outcomes in patients with juvenile localized scleroderma in different pediatric rheumatology clinics in Colombia.
Materials and methodsA descriptive, retrospective, and multicentre study was conducted on patients with juvenile localized scleroderma with a minimum of 1 year of disease onset, and 6 months of follow-up in 10 pediatric rheumatology clinics.
ResultsThe study included 88 patients, with a gender distribution of female 2.1: male 1. Mean age at disease onset was 7.1 years (0–14). Mean disease duration at diagnosis was 16.5 months (1–96). Sub-type distribution was, circumscribed (32.9%), mixed (31.8%), and linear (21.5%), that increased to 55% if linear lesions of the mixed subtype are included), generalized (11.4%), and pan-sclerotic morphea (2.3%). Esthetic compromise was detected in 91%, with growth disturbances in 41%, and joint functional compromise in 32%. Extra-cutaneous involvement occurred in 22.7% and polyautoimmunity in 12.5%.
ConclusionsJuvenile localized scleroderma is a polymorphic and unpredictable disease. It diagnosed late in most of the cases. Extra-cutaneous involvement suggests that is not a disease limited to skin. An early diagnosis, a dynamic treatment and a close follow-up helps to prevent, and detect, complications arising from the disease.
La esclerodermia localizada juvenil es una enfermedad polimórfica que ocurre con mayor frecuencia en niñas. Se acompaña de morbilidad importante. El compromiso extradérmico es frecuente y se reportan tasas de poliautoinmunidad de hasta 7%. Al momento, se desconocen las características clínicas de los pacientes colombianos con esta enfermedad.
ObjetivoDescribir las características clínicas, morbilidades y secuelas en pacientes con diagnóstico de esclerodermia localizada juvenil, en múltiples centros de reumatología pediátrica en Colombia.
Materiales y métodosEstudio descriptivo, retrospectivo y multicéntrico. Pacientes con diagnóstico de esclerodermia localizada juvenil con un mínimo de 1 año de evolución y 6 meses de seguimiento, en 10 centros de reumatología pediátrica mediante revisión de historias clínicas.
ResultadosEl n=88. La distribución por género fue: femenino 2,1; masculino 1. Edad promedio al inicio de la enfermedad 7,1 años (0-14). Promedio de duración de la enfermedad al diagnóstico 16,5 meses (1-96). La distribución por subtipos fue morfea circunscrita (32,9%), mixta (31,8%), linear (21,5%, asciende a 55% al incluir formas mixtas con lesiones lineares) generalizada (11,4%) y panesclerótica (2,3%). Se detectaron alteraciones estéticas en el 91%, alteraciones del crecimiento en 41% y compromiso funcional de articulaciones vecinas en 32%. Se presentó compromiso extradérmico en 22,7% y poliautoinmunidad en 12,5%.
ConclusionesLa esclerodermia localizada juvenil es una enfermedad polimórfica e impredecible. En la mayoría de los casos el diagnóstico es tardío. La tasa de compromiso extradérmico sugiere que no es una enfermedad limitada a la piel. Un diagnóstico temprano, tratamiento dinámico y seguimiento cercano permiten prevenir y detectar tempranamente complicaciones derivadas de la enfermedad.
Scleroderma is an autoimmune, polymorphic disease, characterized by the presence of cutaneous sclerosis secondary to the excessive accumulation of collagen.1,2 It is classified into 2 broad groups, localized scleroderma and systemic scleroderma. It is called localized scleroderma when the skin involvement is not accompanied by affectation of internal organs. In some cases it can involve neighboring structures or even originate distant symptoms, but unlike the systemic form, usually does not compromise vital organs and generates a different morbidity. Systemic scleroderma is characterized by involvement of internal organs and a worse prognosis.1
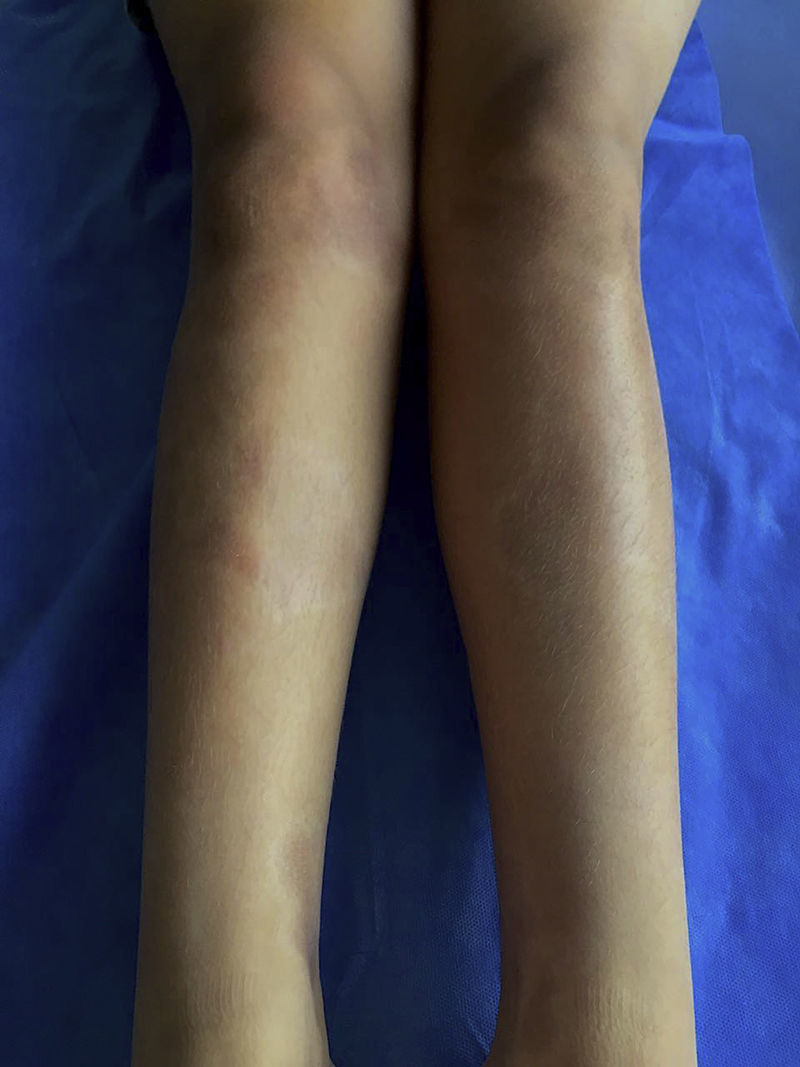
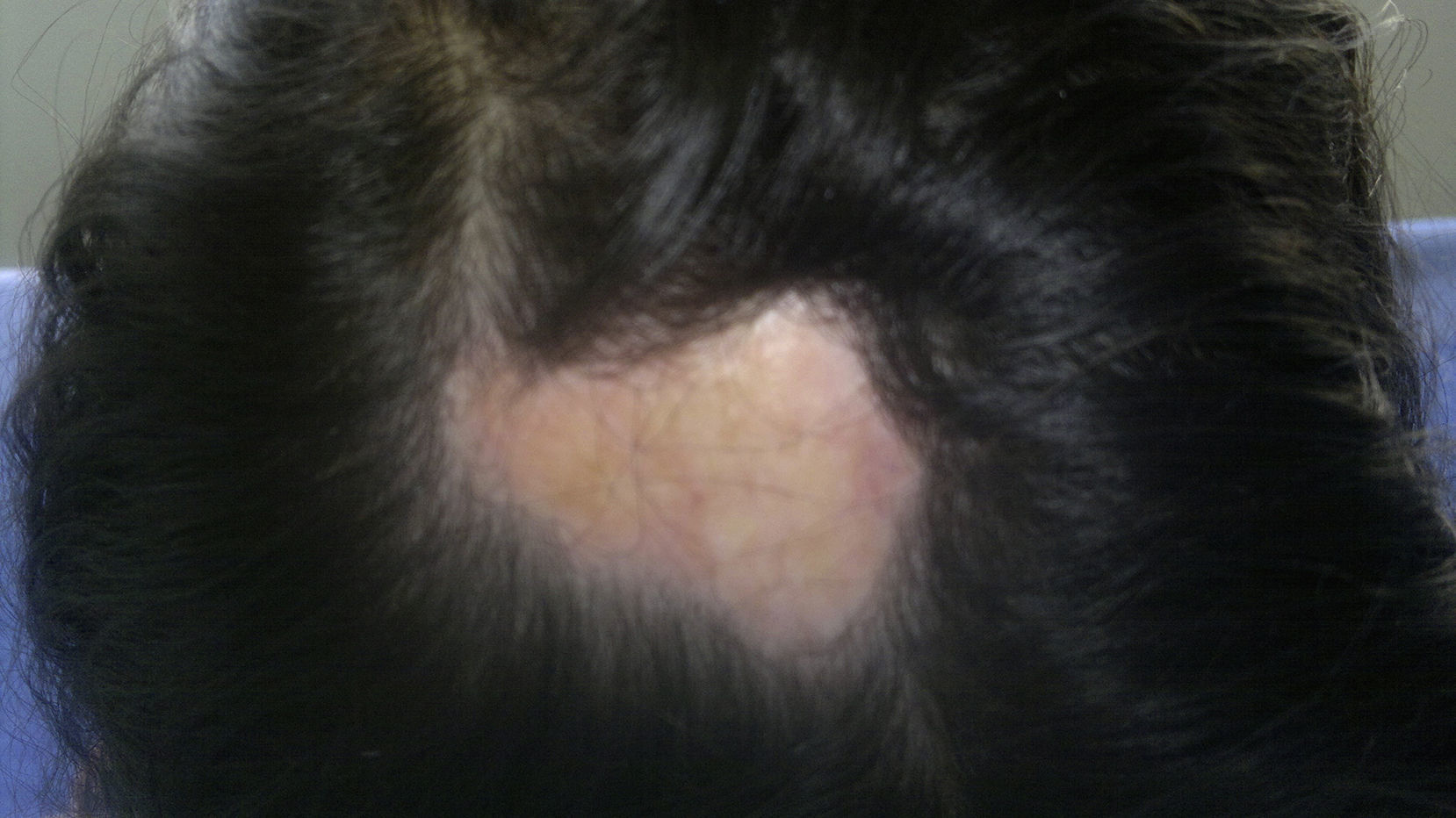
The classification criteria published by the Pediatric Rheumatology European Society (PRes) categorize the juvenile localized scleroderma (jLS) into 5 types according to the skin involvement, they are: linear, circumscribed, mixed, generalized and pan-sclerotic scleroderma. In turn, the circumscribed form is subdivided into superficial and deep forms. Linear scleroderma can compromise the trunk or the limbs (Fig. 1) or be located on the head, where it is called Coup de Sabre (CDS).3 Localized scleroderma affects more frequently the female gender4–8 and its presentation spectrum and clinical course are very varied.

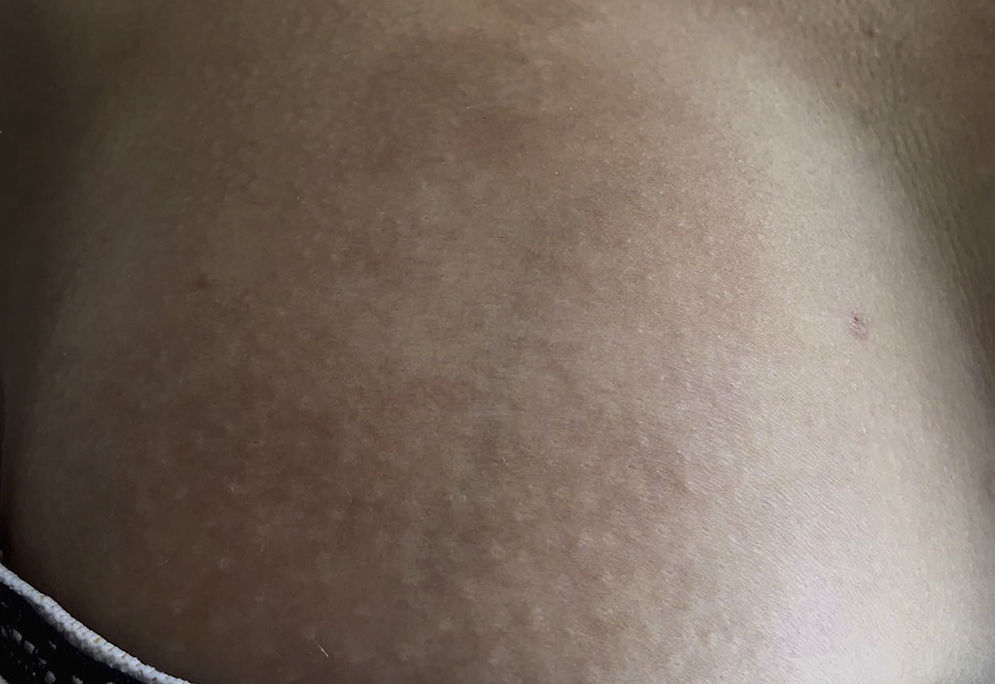
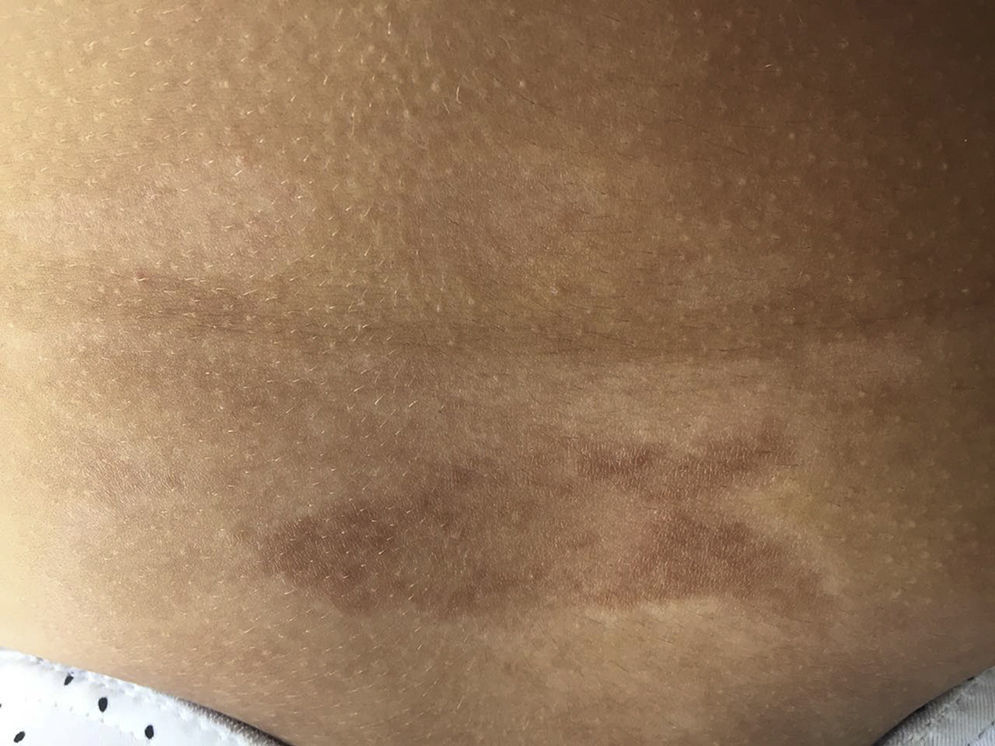
Early lesions (inflammatory phase) are characterized by an erythematosus, violaceous and bright color with a skin thickness that at the beginning is normal (Fig. 2). Over time the fibrosis becomes more prominent with evidence of indurated, hyperpigmented, and in some cases atrophic skin.9 (Fig. 3).


Circumscribed morphea can show improvement with the use of topic treatment; however, close monitoring should be performed given the risk of progression of the lesions and the need to start systemic therapy. Therapeutic options include topical corticosteroids, tacrolimus and imiquimod.9 The use of topical steroids on the face for a long time should be avoided. If upon completion of a maximum of 3 months of topical treatment there is no improvement, systemic therapy must be started.
The other forms of localized scleroderma require a combined management with systemic corticosteroids and disease-modifying anti-rheumatic drugs (DMARDs), for a minimum of 24 months to reduce the risk of relapse. The most widely used DMARD is methotrexate and in case of refractoriness or intolerance to it, mycophenolate mofetil could be used as monotherapy or combined therapy with methotrexate. Other drugs such as intravenous immunoglobulin, infliximab, rituximab, cyclosporine and dapsone have been used in refractory cases with variable responses. Phototherapy has been used as adjuvant therapy due to its antifibrotic and immunosuppressive effect. Effects such as premature aging, carcinogenesis and the lack of effectiveness in deep lesions should always be taken into account.1,2,9–12
The course of localized scleroderma is unpredictable, that is why the early identification of the patients and the beginning of a timely, integral and dynamic treatment are aimed at slowing the progression of the lesions and avoiding complications. The clinical characteristics of the Colombian patients with jLS are so far unknown. Understanding the behavior of the disease in Colombian patients will allow a more specific and timely approach to patients.
The objective of this study is to describe the clinical characteristics, associated morbidities and sequels in a group of patients with definitive diagnosis of jLS followed-up in centers of pediatric rheumatology in the cities of Bogota, Cali, Cartagena and Barranquilla in Colombia.
MethodsA descriptive, retrospective and multicenter study was conducted in a population of patients with a diagnosis of jLS followed-up in 10 centers of pediatric rheumatology in Colombia, by review of medical records. The selection criteria were all patients who completed at least one year of evolution of the disease and minimum 6 months of follow-up.
Given the low prevalence of the disease, we included the universe of patients who met the inclusion criteria.
The variables related to morbidity were the functional commitment of neighboring joints, dyschromia, alopecia and disfiguring lesions, defined when deep alterations were originated in addition to the dyschromic changes, associated or not with growth alterations which were persistent despite treatment. Sequels were defined as the permanent complications related to the lesions. These include growth alterations, which may be of circumferential, longitudinal or mixed type.
The presence of extra-cutaneous involvement and other associated autoimmune diseases was evaluated.
The data were entered in Excel version 15.13.3 and the statistical analysis was performed in the program SPSS version 15.0. The qualitative variables were analyzed by absolute frequencies and percentages, and the quantitative variables were analyzed by means, standard deviations, minimum and maximum. The statistical significance was obtained using the chi-square test.
ResultsA total of 88 patients followed-up in 10 outpatient clinics of pediatric rheumatology who met the inclusion criteria were included.
Demographic characteristicsThe female gender corresponded to 68% of cases. The mean age at the onset of the disease was 7.1 years (0–14 years).
The mean follow-up time was 43 months (6–243 months). The mean duration of the disease at diagnosis was 16.5 months (1–96 months). The mean time between diagnosis and the referral to pediatric rheumatology was 17.1 months (0–156 months). 98% had a biopsy compatible with scleroderma. The 2 remaining patients had lesions on the face very suggestive of this disease and the biopsy was omitted.
According to the PRes classification, the frequency distribution from major to minor was: circumscribed morphea, mixed forms, linear, generalized and pan-sclerotic scleroderma. If the mixed forms that include linear lesions are included, linear scleroderma was the most frequent. The majority of patients had multiple lesions (n=59.67%).
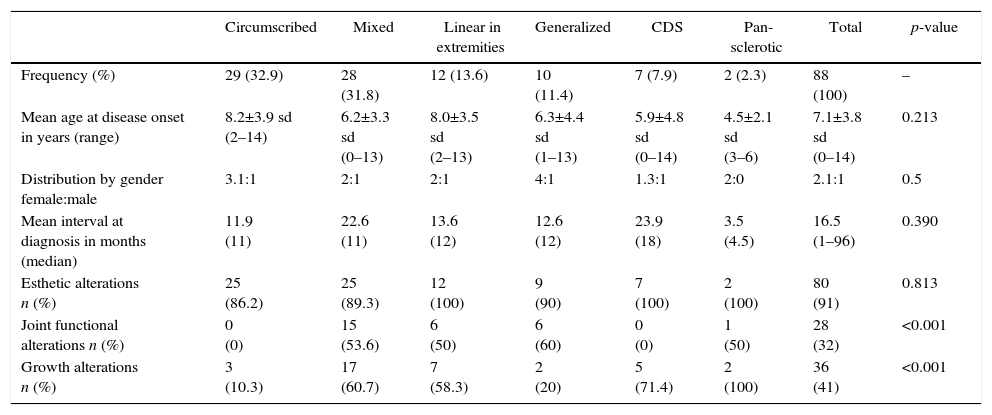
The following morbidities were detected during follow-up: esthetic alterations in 91% of patients, growth alterations in 41% and functional commitment of neighboring joints in 32%. The circumscribed and CDS forms were not associated with functional alterations of neighboring joints. The mixed, linear, CDS and pan-sclerotic subtypes presented the highest rates of growth alterations, being statistically significant. The characteristics according to the PRes classification are summarized in Table 1.
Phenotypes of localized scleroderma according to the PRes classification.
| Circumscribed | Mixed | Linear in extremities | Generalized | CDS | Pan-sclerotic | Total | p-value | |
|---|---|---|---|---|---|---|---|---|
| Frequency (%) | 29 (32.9) | 28 (31.8) | 12 (13.6) | 10 (11.4) | 7 (7.9) | 2 (2.3) | 88 (100) | – |
| Mean age at disease onset in years (range) | 8.2±3.9 sd (2–14) | 6.2±3.3 sd (0–13) | 8.0±3.5 sd (2–13) | 6.3±4.4 sd (1–13) | 5.9±4.8 sd (0–14) | 4.5±2.1 sd (3–6) | 7.1±3.8 sd (0–14) | 0.213 |
| Distribution by gender female:male | 3.1:1 | 2:1 | 2:1 | 4:1 | 1.3:1 | 2:0 | 2.1:1 | 0.5 |
| Mean interval at diagnosis in months (median) | 11.9 (11) | 22.6 (11) | 13.6 (12) | 12.6 (12) | 23.9 (18) | 3.5 (4.5) | 16.5 (1–96) | 0.390 |
| Esthetic alterations n (%) | 25 (86.2) | 25 (89.3) | 12 (100) | 9 (90) | 7 (100) | 2 (100) | 80 (91) | 0.813 |
| Joint functional alterations n (%) | 0 (0) | 15 (53.6) | 6 (50) | 6 (60) | 0 (0) | 1 (50) | 28 (32) | <0.001 |
| Growth alterations n (%) | 3 (10.3) | 17 (60.7) | 7 (58.3) | 2 (20) | 5 (71.4) | 2 (100) | 36 (41) | <0.001 |
The linear form is divided into commitment of the face (CDS) and the extremities.
It was the most common form of scleroderma. It corresponded to one-third of cases. 51.7% (15/29) developed multiple lesions. The most affected body area was the trunk in 44.8% (13/29), followed by the lower limbs in 41.3% (12/29), the face in 17.2% (5/29), the scalp in 13.7% (4/29), the upper limbs in 13.7% (4/29) and the neck in 6.8% (2/29). More than 80% of the patients exhibited dyschromia (82.7%) and 6.8% had alopecia and disfiguring effect. None of the patients had functional joint alterations. 6.8% (2/29) had growth alterations in the upper limbs (one patient with longitudinal commitment and one patient with circumferential commitment). One patient had growth alteration in the lower limbs (longitudinal). Morbidity was related, mostly, with esthetic alterations given the high frequency of dyschromia and to a lesser extent, alopecia and disfiguring effect (Fig. 4).
Mixed scleroderma
One-third of the patients developed mixed forms of the disease. 75% (21/28) corresponded to the association between linear and circumscribed morphea in the extremities. 21.4% (6/28) associated circumscribed morphea and CDS, and one patient (3.6%) presented the association of circumscribed morphea and linear lesions on the extremities and the face (CDS).
The most compromised areas were the trunk and the lower limbs in 75% of patients (21/28), followed by the upper limbs in 50% (14/28), the face in 32.1% (9/28), the neck in 14.2% (4/28) and the scalp in 3.5% (1/28). 78% (22/28) had dyschromic lesions, one-third of the patients had disfiguring lesions (9/28) and 14.2% had alopecia (4/28). More than half of the patients (53.6%) exhibited functional alterations in the joints close to the lesion. Growth alterations located in the lower limbs were observed in 13 patients (46.4%), being in 69% of cases of mixed commitment, longitudinal in 23%, and circumferential in the rest. The rate of growth alterations in the face/scalp was 28.5% (8/28) and in the upper limbs 14.2% (4/28), being in 75% of cases of mixed involvement and circumferential in the rest. This type of scleroderma was associated with multiple complications both esthetic and functional, especially when there were linear lesions associated with lesions of another subtype, multiple lesions or facial involvement.
Linear scleroderma21.5% (19/88) of patients had lesions of linear distribution, but if the patients with mixed forms are included, the frequency rises to 55% of cases (48/88). Seven patients had the variety in CDS and 12 patients had linear lesions in the extremities.
Descriptive analysis according to the subtypes of linear scleroderma:
- -
Scleroderma in CDS:
In addition to the commitment of the face, 71.4% (5/7) associated lesions in the scalp. Multiple lesions were present in 42.8% (3/7) of cases. All patients developed esthetical alterations given by the presence of dyschromic lesions 85.7% (6/7), disfiguring lesions in 85.7% (6/7) and an additional 57% developed localized alopecia (4/7). Five patients (71.4%) had growth alterations in the face/skull. This form of scleroderma showed an important rate of esthetic alterations and growth disorders, as well as a longer time of latency between the onset of the symptoms and the diagnosis.
- -
Linear scleroderma on the trunk and/or the extremities:
The lesions were located in the same proportion in the upper and lower limbs (50% and 50%). There was no trunk involvement. The majority of the patients had single lesions (10/12, 83%). All patients had esthetic alterations due to dyschromia. Half of them (6/12) presented functional involvement of neighboring joints and growth alterations in the extremities (3/12 in the upper limbs and 3/12 in the lower limbs). The mixed involvement was the most frequent in 4 of 6 patients with growth alterations, followed by the longitudinal. One patient required epiphysiodesis of the left distal femur and proximal tibia for the correction of discrepancy in length of the lower limbs and enlargement of the extensor hallucis and the right extensor digitorum longus due to a claw deformity of the ipsilateral second toe. The morbidity of this subtype of scleroderma, in addition to the esthetic alterations, was associated to a significant rate of growth alterations (longitudinal, circumferential or mixed) and functional commitment of the joints near the lesion.
All patients with this form of scleroderma exhibited commitment of the trunk and lower limbs. Commitment of the upper limbs was present in 80% of cases (8/10), of the neck in 60% (6/10) and of the face in 40% (4/10). There were no lesions on the scalp. The esthetic commitment was important with the presence of dyschromic lesions in almost all patients (n=9, 90%) and disfiguring effect in 20% (n=2). More than half of the affected individuals developed functional commitment of the neighboring joints (n=6, 60%) and one fifth of the patients had growth alterations (2/10, one in the face and the other in the upper and lower limbs with longitudinal involvement). One patient had calcinosis, which was characterized by being a form with extensive skin lesions, an important rate of growth alterations and it was the form of scleroderma with the highest frequency of functional commitment of neighboring joints.
Pan-sclerotic sclerodermaIt was present in 2 female patients. One patient had articular involvement given by arthritis and polyarticular functional commitment. Both presented multifocal esthetic alterations (extremities, trunk and face) and growth disorders in the limbs (mixed). One of them required amputation of the left lower limb secondary to severe septic arthritis of the knee. It was characterized by being the most severe and progressive form that caused important esthetic and functional commitment.
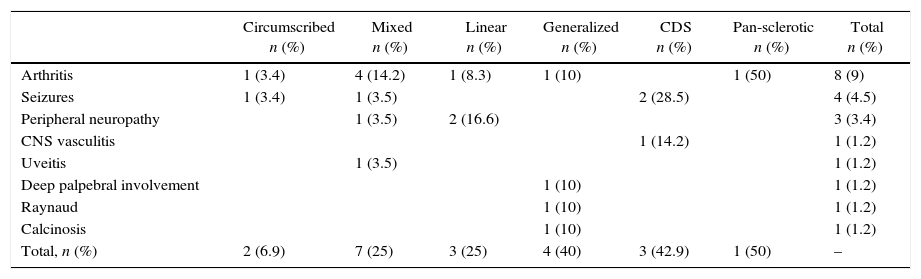
Extra-cutaneous involvementIt was detected in a quarter of patients (n=20, 22.7%). The type with the highest frequency of extra-cutaneous involvement was the CDS in 42.9% followed by generalized scleroderma in 40% of patients. The linear form was the most frequently associated to articular involvement (in pure or mixed forms). The affection of the central nervous system occurred more frequently in patients with lesions that involved the face/skull (Table 2).
Extra-cutaneous involvement according to the types and subtypes of scleroderma.
| Circumscribed n (%) | Mixed n (%) | Linear n (%) | Generalized n (%) | CDS n (%) | Pan-sclerotic n (%) | Total n (%) | |
|---|---|---|---|---|---|---|---|
| Arthritis | 1 (3.4) | 4 (14.2) | 1 (8.3) | 1 (10) | 1 (50) | 8 (9) | |
| Seizures | 1 (3.4) | 1 (3.5) | 2 (28.5) | 4 (4.5) | |||
| Peripheral neuropathy | 1 (3.5) | 2 (16.6) | 3 (3.4) | ||||
| CNS vasculitis | 1 (14.2) | 1 (1.2) | |||||
| Uveitis | 1 (3.5) | 1 (1.2) | |||||
| Deep palpebral involvement | 1 (10) | 1 (1.2) | |||||
| Raynaud | 1 (10) | 1 (1.2) | |||||
| Calcinosis | 1 (10) | 1 (1.2) | |||||
| Total, n (%) | 2 (6.9) | 7 (25) | 3 (25) | 4 (40) | 3 (42.9) | 1 (50) | – |
CNS: central nervous system.
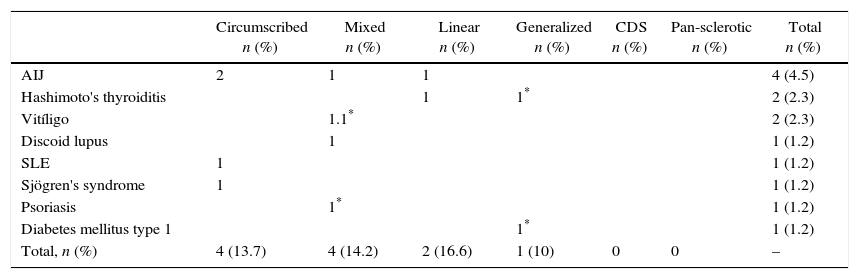
Eleven patients developed another autoimmune disease during follow-up (12.5%). The most prevalent was juvenile idiopathic arthritis followed by Hashimoto's thyroiditis and vitiligo. The distribution by subtypes of juvenile idiopathic arthritis was: 2 patients with the oligoarticular form, one with polyarticular and one with enthesitis-related arthritis. Two patients presented multiple autoimmune disease defined as 3 or more autoimmune diseases. One of them with localized scleroderma, vitiligo and psoriasis, confirmed by biopsy in that order of appearance. Another patient associated diabetes mellitus type 1, localized scleroderma and Hashimoto's thyroiditis, in that order of appearance (Table 3).
Presence of polyautoimmunity according to the types and subtypes of scleroderma.
| Circumscribed n (%) | Mixed n (%) | Linear n (%) | Generalized n (%) | CDS n (%) | Pan-sclerotic n (%) | Total n (%) | |
|---|---|---|---|---|---|---|---|
| AIJ | 2 | 1 | 1 | 4 (4.5) | |||
| Hashimoto's thyroiditis | 1 | 1* | 2 (2.3) | ||||
| Vitíligo | 1.1* | 2 (2.3) | |||||
| Discoid lupus | 1 | 1 (1.2) | |||||
| SLE | 1 | 1 (1.2) | |||||
| Sjögren's syndrome | 1 | 1 (1.2) | |||||
| Psoriasis | 1* | 1 (1.2) | |||||
| Diabetes mellitus type 1 | 1* | 1 (1.2) | |||||
| Total, n (%) | 4 (13.7) | 4 (14.2) | 2 (16.6) | 1 (10) | 0 | 0 | – |
SLE: systemic lupus erythematosus.
Autoantibodies were detected in 46% of patients. Antinuclear antibodies were detected in 42%. ENAS were positive in patients with associated SLE or Sjögren's syndrome and in 2 patients without another associated autoimmune disease. Anti-DNA was present in the patient with SLE. Rheumatoid factor was detected in 2 patients, one of them developed arthritis as extra-cutaneous involvement of the scleroderma. Two patients with Hashimoto's thyroiditis had positive antithyroid antibodies. Two patients had positive anti-SCL70. Anticardiolipin IgG and lupus anticoagulant were present, each in one patient. None of the patients developed clinical signs or serology compatible with systemic sclerosis during the follow-up.
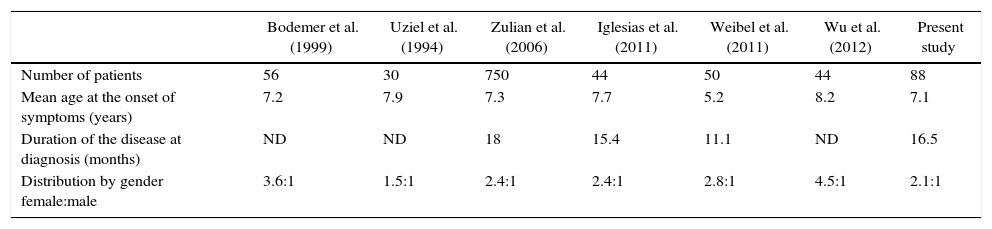
DiscussionThis study represents the first characterization of a group of Colombian patients with jLS. The studies report a higher frequency in girls4–7 with an age of onset of symptoms around 7 years (Table 4), being consistent with the findings of this study.4–6,8,11,13 The predominance in the female gender was more marked in generalized scleroderma, with a gender distribution: female:male of 4:1.
Comparative chart between different pediatric series of localized scleroderma.
| Bodemer et al. (1999) | Uziel et al. (1994) | Zulian et al. (2006) | Iglesias et al. (2011) | Weibel et al. (2011) | Wu et al. (2012) | Present study | |
|---|---|---|---|---|---|---|---|
| Number of patients | 56 | 30 | 750 | 44 | 50 | 44 | 88 |
| Mean age at the onset of symptoms (years) | 7.2 | 7.9 | 7.3 | 7.7 | 5.2 | 8.2 | 7.1 |
| Duration of the disease at diagnosis (months) | ND | ND | 18 | 15.4 | 11.1 | ND | 16.5 |
| Distribution by gender female:male | 3.6:1 | 1.5:1 | 2.4:1 | 2.4:1 | 2.8:1 | 4.5:1 | 2.1:1 |
ND: no data.
In the series of Zulian et al.,4 Uziel et al.5 and Wu et al.,13 linear commitment is reported as the most frequent, according to what was presented in this study.4,5,13 As for the distribution by types of scleroderma, it should be considered that given the differences in the classification of the entity, it is difficult to compare findings of the different studies.
Although scleroderma causes lesions whose characteristics of brightness, induration and dyschromia should favor early diagnosis, a high proportion of patients with late diagnosis is observed in all series (Table 4).4–6,8,14,15
It is surprising that in this series, the mixed forms and CDS presented the longest times of latency between onset of symptoms and diagnosis, despite the multiplicity of lesions that characterize the generalized forms and the evident lesions on the face in CDS. This delay in the diagnosis may be related to late consultations, lack of recognition of the disease by the physician or late referral to dermatology and rheumatology.
The lesions determined esthetic alterations in 91% of cases. These were classified as dyschromia, alopecia and disfiguring effect. The lesions of scleroderma of linear, mixed, generalized and pan-sclerotic subtypes caused functional alterations of the neighboring joints in more than half of the cases.
Localized growth alterations developed in 58, 60, 71.4 and 100% of patients with linear, mixed, CDS and pan-sclerotic forms, respectively. Uziel et al.,5 report a rate of growth alterations of 26% in patients with linear scleroderma in the extremities.5 Zulian et al.,4 report atrophy of one limb with autoamputation in one patient with the pan-sclerotic form4 and Wu et al.,13 report shortening of the extremities in 5 patients.13 The present study describes the commitment of growth in the different types and subtypes of scleroderma.
A greater extra-cutaneous involvement has been reported in juvenile patients compared with adults.16 Gorkiewicz et al.,17 report a rate of extra-cutaneous involvement of 24–64% of patients with localized scleroderma in adults and children (average 20%), being higher in the linear and generalized forms.17 22.4% of patients in the series of Zulian et al.,18 which only included juvenile patients (n=750), developed extra-cutaneous involvement, with a frequency similar to that observed in this series (22.7%).18
Arthritis was the most frequent clinical manifestation of extra-cutaneous involvement. As reported by Zulian et al.,18 it occurred most frequently in the linear commitment of the limbs. Neurological involvement, characterized by seizures, peripheral neuropathy and vasculitis of the CNS, is the second most frequent. The last patient was clinically characterized by headache. As in the series described, neurological involvement was more prevalent in patients with lesions on the face.18
The case of uveitis occurred in a patient with CDS. The frequency of ocular involvement was lower than that reported in the literature.14,18 This difference could be determined by an underdiagnosis, since a regular ophthalmologic evaluation was not conducted in all patients with lesions on the face/scalp.
Neurological and ocular involvement in patients with lesions on the face/scalp suggests the need for a regular neurological and ophthalmologic evaluation in these patients.
12.5% presented polyautoimmunity. This rate was higher than the 7% reported by Zulian et al.,18 suggesting the need to raise the level of alert to the appearance of other autoimmune diseases.
At the time of the follow-up there was no progression of the disease to systemic sclerosis in any patient.
The limitations of the study include the absence of information on a family history of autoimmunity and the treatment received by the patients, and the fact that a standardized follow-up tool was not used in the patients.
In conclusion, this study allows the characterization of jLS in a group of Colombian patients and demonstrates that it is not a disease limited to the skin. Despite advances in this subject, in most cases the diagnosis is established late. Circumscribed scleroderma is accompanied by important esthetic alterations, while the linear, mixed, generalized and pan-sclerotic forms are also accompanied by important functional alterations and localized growth disorders. The associated morbidity is accompanied by a negative and permanent impact on the quality of life of the patients with a diagnosis of jLS.
ConclusionsjLS is a polymorphic and unpredictable disease. It determines an important morbidity. Late diagnosis is frequent in children and adults, and may be accompanied by a negative impact on the prognosis by allowing the progression of the lesions in size, number and depth. Extra-cutaneous involvement was present in almost a quarter of patients, which reflects that it is not a disease limited to the skin.
The presence of polyautoimmunity was observed in a non-negligible frequency and 2 patients developed multiple autoimmune disease. The possible deregulation of the immune system as an important part in the pathogenesis of the disease is confirmed. An early diagnosis, a dynamic treatment and a close follow-up can help to prevent and detect early complications derived from the disease. It is necessary to raise awareness among the medical staff of the non-benign nature of this condition. Learning, early recognition of this pathology and a timely referral to the rheumatologist should be promoted. Likewise, the understanding of the disease by the patient and caregivers, the importance of adherence to treatment and follow-up and the early detection of complications should be ensured in order to improve the prognosis in the patients diagnosed with jLS.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare they do not have any conflict of interest.
Please cite this article as: Arango C, Malagón C, del Pilar Gómez M, Mosquera C, Yépez R, González T, et al. Esclerodermia localizada juvenil: ¿es una enfermedad benigna? Rev Colomb Reumatol. 2017;24:145–152.
