


Anderson–Fabry disease is the second most common lysosomal storage disease after Gaucher disease. It is an X-linked lysosomal disorder that causes a deficiency in alpha-galactosidase, leading to the accumulation of globotriaosylceramide (Gb3) in the lysosomes of different cells, producing renal, cardiac and neurological deficits that can lead to an early death. The renal histologic analysis, applied using a standardised rating system, is useful for initiating enzyme replacement therapy and for assessing the prognosis. In this article we classify the histologic lesions found in light and electron microscopy in renal biopsies of patients with Anderson–Fabry disease, using the scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). More than half of the cases did not present any change in the clinical and laboratory assessment at the time of the biopsy; nevertheless there were changes in the light and electronic microscopy findings. The information from the renal biopsy is an early indicator of renal damage, even without proteinuria and with preserved renal function.
La enfermedad de Fabry-Anderson es la segunda enfermedad de depósito lisosomal más frecuente después de la enfermedad de Gaucher. Es un desorden ligado a X, que causa deficiencia de α-galactosidasa, dando la acumulación de globotriaosilceramida (Gb3) dentro de los lisosomas de diversas células, lo que conduce a un deterioro renal, cardiaco y neurológico, que puede llevar a la muerte temprana. El análisis histológico renal, aplicado a un sistema de puntuación estandarizado, es útil para el inició de la terapia de sustitución enzimática y la evaluación del pronóstico. En este trabajo se clasificaron las lesiones histológicas encontradas en la microscopía de luz y la microscopía electrónica, en biopsias renales de pacientes con enfermedad de Fabry-Anderson, utilizando el Sistema de Puntuación de Patología Renal en Enfermedad de Fabry: Informe del Grupo Internacional de Estudio en Nefropatía de Fabry (ISGFN). Más de la mitad de los casos no presentaban alteraciones en la evaluación clínica y de laboratorio al momento de la biopsia, aun así, se encontraron cambios en la microscopía de luz y en la electrónica. La información proporcionada a través de la biopsia renal es un indicador temprano de daño renal, incluso sin datos de proteinuria y con función renal conservada.
The metabolic defect in Fabry disease is a deficiency in the lysosomal hydrolase alpha-galactosidase A (alpha-Gal A), characterised by the hydrolytic excision of globotriaosylceramide (Gb3) and lyso-globotriaosylsphingosine (lyso-Gb3). Both Gb3 and lyso-Gb3 are elevated in male patients with classic Fabry disease, but may be only slightly increased or normal in late-onset or heterozygous variants.8 The protein alpha-Gal A is coded by a gene at 12-kb on the long arm of the X chromosome (Xq22.1).9 More than 400 mutations have been identified.10,11 Most families have specific or private mutations and de novo mutations are rare.6
The disease's prevalence is estimated at 1:17,000 to 1:117,000 in Caucasian males. However, it is found in all ethnic and racial groups.2,3 All males with the enzyme defect develop the disease, while female carriers are normally asymptomatic or express an attenuated form.4,5 Kidney failure was the principal cause of death prior to the advent of dialysis treatment and the mean age of death was 41 years. Nowadays, dialysis and pharmacological antiproteinuric treatment have extended this to 55 years in 50% of homozygous patients.6
The Fabry Outcome Survey (FOS) and the Fabry Registry are international databases of patients with Fabry disease. An analysis of both reveals that the most common cause of death in 2001 was renal failure. In a recent study from 2001 to 2007, the authors reported that the most common cause of death was cardiovascular, at approximately 34% of male sufferers and 57% of female sufferers. This change in the principal cause of death appears to be due in particular to renal replacement therapy.7
The diagnosis is generally established by measuring alpha-Gal A activity in plasma or leukocytes in peripheral blood samples, fibroblast cultures, or by using dried blood spot samples on filter paper.13 On the other hand, a diagnosis may initially be made through a renal biopsy taken to assess another type of chronic kidney disease (nephrotic syndrome, haematuria or other symptoms).14,15 Nevertheless, although the histology may suggest the disease (zebra bodies), cases have been reported in which the changes observed under light and electron microscopy were indistinguishable from Fabry disease, but the lesions were caused by drugs (chloroquine, hydroxychloroquine, chlorphentermine, chlorcyclizine, imipramine, clomipramine, gentamicin and amiodarone), wherein the distinction was made using alpha-Gal A measurements.
The accumulation of Gb3 in the kidneys occurs first and foremost in the glomeruli (podocytes, endothelial and mesangial cells) as well as in the distal tubule. Morphological assessment using light and electron microscopes has revealed that glomerular and vascular changes occur in spite of normal kidney function.16
Glomerulosclerosis and fibrosis of the interstitial tubule are the histological characteristics that correlate most strongly with the progression of renal involvement, which translates to a need for renal replacement therapy (RRT). Kidney failure increases the risk of cardiovascular events.1,7
The drop in kidney function is related to the degree of proteinuria in untreated patients. Male sex and hypertension are also significant risk factors for the development of kidney failure.17 As with any nephropathy, the protein overload can cause an increase in the levels of inflammatory mediators and interstitial accumulation of these mediators can lead to scarring of the kidneys.18
Proteinuria is an early sign of Fabry disease in both sexes and is the most frequent clinical manifestation.2 Among patient in the FOS database, proteinuria was present in 44–54% of males and 33–41% of females. Similarly, the Fabry Registry recorded frank proteinuria (>300mg/day) in 43% of males and 26% of females with stage I CKD; the rate was even higher in patients with more advanced renal involvement.3
A kidney biopsy is useful in all patients with any degree of proteinuria, albuminuria and/or renal dysfunction to assess the degree of glomerulosclerosis and interstitial damage due to its prognostic significance. In patients with minimal proteinuria and normal kidney function, a biopsy can also determine whether there are significant deposits of Gb3 (especially in podocytes and endothelial cells), reveal early damage indicating enzyme replacement therapy (ERT), or provide prognostic evidence, depending on the degree of glomerular sclerosis.19 A biopsy may also be performed if there is the possibility of co-existing pathologies (for example, diabetes of other glomerular diseases, such as IgA nephropathy or thin basement membrane disease) and if there is a sudden inexplicable drop in kidney function7.
An international study systematically classified renal histologic lesions in patients with Anderson–Fabry disease using the SCORING SYSTEM FOR RENAL PATHOLOGY IN FABRY DISEASE: REPORT OF THE INTERNATIONAL STUDY GROUP OF FABRY NEPHROPATHY (ISGFN).20 The group was made up of 10 reference centres for Fabry disease and included a total of 35 male and 24 female patients, the majority with mild clinical nephropathy. Various histologic aspects were assessed and a morphometric index of chronic damage was obtained for each biopsy. The outcome was a validated scoring system, developed with the long-term objective of being able to determine whether basic histologic information may be related to the rate of progression and/or response to enzyme replacement therapy in Fabry nephropathy.
The results of the study were as follows:
- -
Mild or severe segmental sclerosis was observed in 19 out of 35 males and 11 out of 24 females. The average degree of segmental sclerosis was 4.7±7.8% glomeruli. Global sclerosis was present in 14.8±22.7% glomeruli.
- -
Interstitial fibrosis was observed in all biopsies.
- -
Females had significantly fewer podocytic inclusions than males, and one female subject had no inclusions.
- -
Tubular inclusions were present in the distal tubule in 42 of 56 cases (75%). Inclusions in the proximal tubule were more common in males than in females (47.1% versus 19%). Inclusions in both the proximal and distal tubules were present in 14 male and 4 female subjects.
- -
Capillary and arteriolar inclusions were identified in 26 male and 7 female subjects. Inclusions in the vascular intima were more frequent in males than in females. Hyalinosis was present in the arterioles of 34 patients and in the arteries of 22 patients out of the 55 assessed.
The authors concluded that glomerulosclerosis and interstitial fibrosis are found in both sexes, including in patients with minimal or no proteinuria. These two data points develop early in the disease and the absence of clinical signs of kidney damage does not rule out Fabry nephropathy.
Although no significant differences were found between male and female patients in segmental and global sclerosing lesions, they could be an early prognostic indicator or chronic kidney disease.
The clinical disease was milder in women, with less global sclerosis and fewer podocytic, vascular and proximal tubule inclusions in comparison to men. However, there were no significant differences between the sexes with regard to segmental sclerosis, interstitial fibrosis, inclusions in distal tubules or arteriolar hyalinosis.20
Enzyme replacement therapy (ERT) with agalsidase-beta (recombinant human alpha-galactosidase A) removes Gb3 from the vascular endothelium in patients with Fabry disease.14
In Mexico, the use of ERT is currently recommended by some public healthcare systems based on evidence that the use of ERT safely and effectively reduces plasma and urine levels of GL-3, maintains stable kidney function and eliminates Gb3 deposits from the renal vascular epithelium.21
Patients and methodsThis is an observational, descriptive, retrospective cross-sectional study. It includes all renal biopsies of patients with Anderson–Fabry disease received at Hospital General de México from 2009 to 2015. Renal biopsies with results reported as a biopsy unsuitable for diagnosis and cases lacking an electron microscopy report were excluded. Selected cases in which a diagnosis was not reached or in which the study data were incomplete were eliminated.
The data collection was performed as follows: The first step consisted of collecting information on all renal biopsies received between 2009 and 2015 at Hospital General de México, selecting those with a clinical diagnosis of Anderson–Fabry disease; the inclusion and exclusion criteria would be used to select the cases for the study. All of the histopathological diagnoses for these cases were then collected, as well as the electron microscopy study and clinical information. A database was created with the resulting information. Descriptive statistical analyses were performed using non-parametric techniques.
The confidentiality of the data is one of the most important ethical aspects. The personal data of the patients whose renal biopsies were used in the study were neither used nor known.
ResultsAll renal biopsies received between 2012 and 2015 at Hospital General de México were reviewed in the surgical pathology department's database, selecting those entered with a confirmed diagnosis of Anderson–Fabry disease. We then reviewed the light microscopy and electron microscopy studies and the information based on the SCORING SYSTEM FOR RENAL PATHOLOGY IN FABRY DISEASE: REPORT OF THE INTERNATIONAL STUDY GROUP OF FABRY NEPHROPATHY (ISGFN) and created a database with the resulting information.
Of a total of 2500 renal biopsies received over a four-year period (from 2012 to 2015), 20 were cases with a confirmed diagnosis of Anderson–Fabry disease. The distribution according to sex was 40% male (8 cases) and 60% female (12 cases).
The mean age was 18.1±11.6 years with a minimum age of 4 years and a maximum of 43. The mean age of the male subjects was 20.6±12.4 years and 16.4±11.3 years for female subjects.
Proteinuria in urine was elevated in seven cases (35%), creatinine in two (10%) and a further two cases had decreased glomerular filtration (10%).
With regard to histological findings in the light microscopy study, we obtained the following data: Glomerulosclerosis. Focal global sclerosis was observed in four cases (20%), three male and one female. Segmental sclerosis was present in 12 cases (60%), five male and seven female. Two cases with global sclerosis showed elevated proteinuria, as well as half of the cases with segmental sclerosis (6 cases).
Five cases were found to have some degree of fibrosis (25%). Three of these also had increased proteinuria and two had elevated creatinine and a decreased GFR.
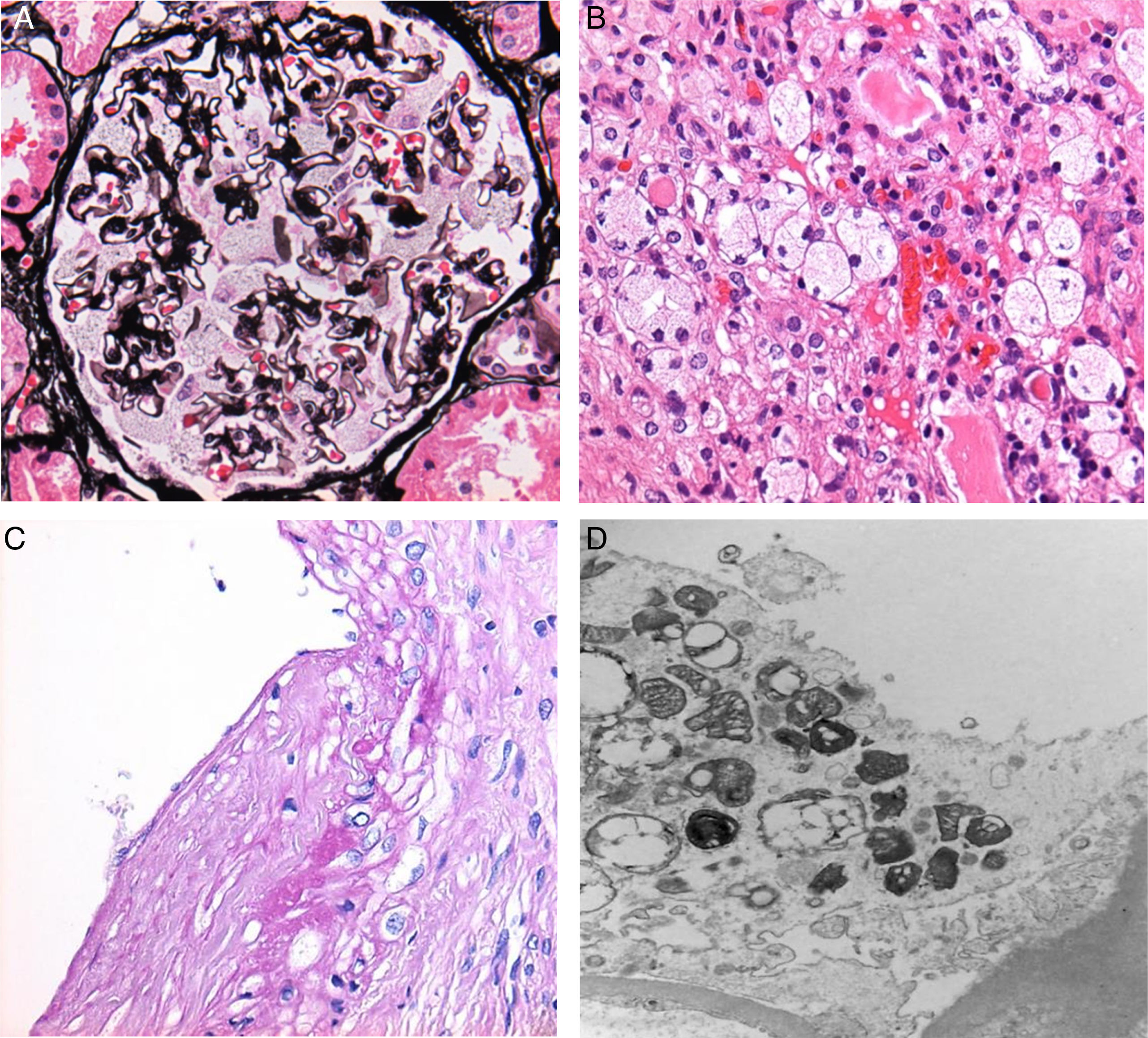
Adhesions between the capillary tufts and Bowman's capsules were observed in 15 cases (75% of all cases). Podocytic inclusions were found in all of the biopsies analysed (Fig. 1A).
 Vacuolation of podocytes. (B) Tubular and interstitial deposits. (C) Deposits in blood vessels. (D) EM: zebra bodies in the cytoplasm of podocytes.")
Arteriolar sclerosis (mild/moderate/severe) was found in 13 renal biopsies (65% of all cases).
With regard to findings in the electron microscopy study, we obtained the following data: Podocytic inclusions in 19 biopsies (zebra bodies) (Fig. 1D). The only case in which these inclusions were not found was that of a male patient who presented global and segmental lesions, proteinuria, elevated creatinine and decreased GFR, with a confirmed cytogenetic and clinical diagnosis of Anderson–Fabry disease.
Tubular inclusions were observed in 14 cases (70%) and vascular inclusions in 11 (55%); these findings were also visible under light microscopy (Fig. 1B and C).
DiscussionThe mean age encountered in this study (18.1±11.6 years) was lower than that reported in the literature (39.4±13.2 years), most likely due to the fact that the study population corresponded to family groups that included minors.
It was not possible to unify the proteinuria, creatinine and GFR values due to the variation in ages (4–43 years), as different formulae were used to calculate GFR, proteinuria and creatinine; most of the biopsies also came from other hospitals, with different reference values for each laboratory. Nevertheless, the decision was made to evaluate these variables, taking as positive those cases reported as subnephrotic proteinuria, nephrotic syndrome, elevated creatinine and decreased GFR.
Proteinuria is reported as the earliest clinical sign in Fabry disease (in 44–54% of male patients and in 33–41% of female patients). In this study, it was found in 35% of cases, which is slightly lower than that reported in the international literature.
In one case no inclusions (zebra bodies) were found in the electron microscopy study. Similar cases have been reported in the literature and this does not rule out a diagnosis of Fabry disease or absence of renal involvement.
The most common histological changes found were segmental sclerosis, early adhesions and podocytic inclusions (zebra bodies), which were visible under both light and electron microscopy, even in patients with minimal or no detected alterations in the clinical and laboratory assessments (e.g. proteinuria). These two data points are supported by the literature, where they are mentioned as prognostic indicators with the recommendation to initiate enzyme replacement therapy (ERT).
In view of the above, we conclude that the information provided through the renal biopsy in nephropathy associated with Fabry disease is an early indicator of renal damage, even without proteinuria and with preserved renal function.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare that they have no conflict of interests.