



Hepatic involvement in primary amyloidosis is an infrequent challenge to the hepatologist. Although usually asymptomatic, amyloidosis may have unusual manifestations. Liver biopsy is an important diagnostic tool for this condition. Herein, we report three cases of portal hypertension related to primary hepatic amyloidosis, one of them in the form of acute liver failure.
Amyloidosis is a generic term that refers to the extracellular tissue deposition of fibrils composed of low molecular weight subunits (5 to 25 kD) of a variety of normal serum proteins, which stain with Congo red. It may be classified as primary (light chain-related, AL amyloidosis), which is the most common type, or secondary (generally associated to chronic infections, inflammatory or neoplastic disorders, AA amyloidosis). Other less frequent forms of amyloidosis include familial (associated with positive transthyretin), senile, localized (skin, bladder and other organs), and dyalisis-related (associated with β2-microglubulin).1 Hence, serum free light chain analysis differentiates AL amyloidosis from other forms of amyloidosis. AL is the main type of amyloidosis in the United States2 (AA amyloidosis being the most common one worldwide) and is frequently associated with underlying plasma cell dyscrasias. Several organs may be affected, such as kidney, heart, autonomic and peripheral nervous system, muscle and skin, among others. Liver involvement is common and could be observed in around 70% of the cases in one autopsy series.3 Nevertheless, hepatic deposits of amyloid rarely cause clinical manifestations.4 Of note, portal hypertension (PH) and acute liver failure (ALF) are two particularly uncommon manifestations of AL amyloidosis.5–7 Herein we report three cases of hepatic AL amyloidosis with unusual and challenging clinical presentations.
Case ReportCase 1A 72 year-old woman with hypertension was admitted to her local hospital with a history of two months of malaise, anorexia, weight loss and ascites (Table 1). There was no record of alcohol or drug abuse. Besides ascites, physical examination was unremarkable. An abdominal ultrasound revealed an enlarged liver, a modest amount of ascites, normal suprahepatic veins caliber, a normal spleen and normal kidneys. Results of laboratory blood and urine tests were as it follows (normal range in brackets): alkaline phosphatase (ALP) 619 U/L (30-100 U/L), gammaglutamyltransferase (GGT) 399 U/L (4-50 U/L), aspartate aminotransferase (AST) 125 U/ L (9-25 U/L), alanine aminotransferase (ALT) 128 U/L (7-30 U/L), total bilirubin 1.13 mg/dL (0-1 mg/dL), albumin 3.3 g/dL (3.5-5 gr/dL), INR 1.2 (0.8-1.2), creatinine 1.9 mg/dL (0.5-0.9 mg/dL), estimated glomerular filtration rate: 28 mL/min/1.73 m2, uric acid 5.6 mg/dL (2.3-6.6 mg/dL), hematocrit 47.9% (36-46%), white cell count 6.400 103/ mm3 (4.5-11), platelet count 162 103/mm3 (140-400), there were Howell-Jolly ++ bodies in the peripheral blood smear, erythrocyte sedimentation rate: 2 mm/ h, urinary protein 352 mg/24 h (< 150 mg/24 h). Serum markers of viral hepatitis B and C as well as antinuclear antibodies, anti-smooth-muscle antibodies and anti-mitochondrial antibodies were negative. Ascitic fluid analysis showed total protein concentration 0.6 g/dL, serum to ascites albumin gradient (SAAG) of 2.1 g/dL (compatible with PH-dependent ascites).
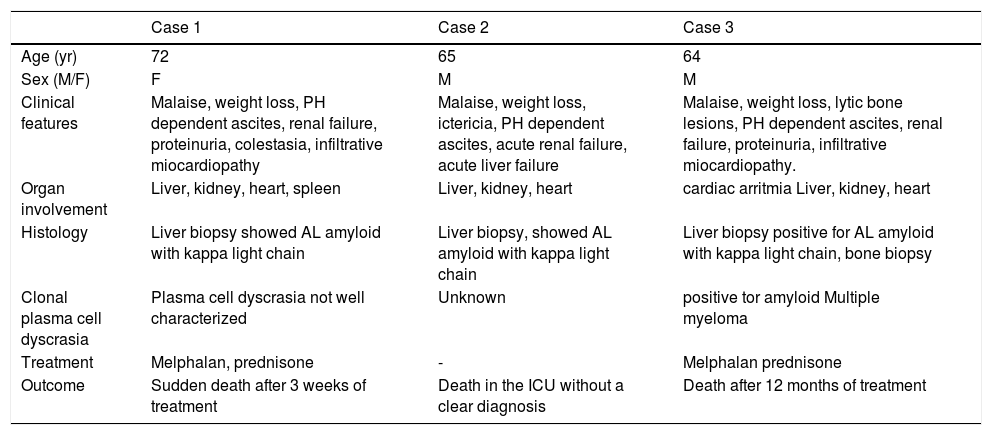
Demographic data, main clinical manifestations, laboratory, liver images and echocardiogram in the three patients.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age (yr) | 72 | 65 | 64 |
| Sex (M/F) | F | M | M |
| Clinical features | Malaise, weight loss, PH dependent ascites, renal failure, proteinuria, colestasia, infiltrative miocardiopathy | Malaise, weight loss, ictericia, PH dependent ascites, acute renal failure, acute liver failure | Malaise, weight loss, lytic bone lesions, PH dependent ascites, renal failure, proteinuria, infiltrative miocardiopathy. |
| Organ involvement | Liver, kidney, heart, spleen | Liver, kidney, heart | cardiac arritmia Liver, kidney, heart |
| Histology | Liver biopsy showed AL amyloid with kappa light chain | Liver biopsy, showed AL amyloid with kappa light chain | Liver biopsy positive for AL amyloid with kappa light chain, bone biopsy |
| Clonal plasma cell dyscrasia | Plasma cell dyscrasia not well characterized | Unknown | positive tor amyloid Multiple myeloma |
| Treatment | Melphalan, prednisone | - | Melphalan prednisone |
| Outcome | Sudden death after 3 weeks of treatment | Death in the ICU without a clear diagnosis | Death after 12 months of treatment |
A liver biopsy was performed evidencing altered hepatic parenchyma due to deposits of an amorphous, eosinophilic, extracellular material that stained with Congo red dye and appeared apple green under polarized light consistent with amyloid. The patient was then sent to our liver unit. At our hospital ascites was treated with diuretics, but severe hyponatremia developed, so the treatment had to be interrupted. The liver biopsy was re-examined. Immunohistochemical staining was positive for k light chains. The final diagnosis was AL amyloid with êappa light chain positivity. Bone marrow smear showed 15% of plasma cells. Flow citometry from bone marrow aspirate showed two populations of plasma cells; 12% of normal plasma cells and 88% of clonal plasma cells that had positive intracellular kappa light chain stain. She had normal serum protein electrophoresis and normal urine immunofixation (proteins 1.53 g/24 h, with no monoclonal component); normal serum calcium (9.1 mg/dL normal value 8.5-10.5 mg/dL), serum immunoglobulin (Ig) G 1,990 mg/dL (694-1,618 mg/dL), IgA 312 mg/dL (68-378 mg/dL), IgM 257 mg/dL (60-263 mg/dL). Skeletal survey was incomplete; she only had a normal chest X-Ray. She never showed anemia. Renal function worsened after diuretic use. There was no bone marrow biopsy.
Echocardiogram showed severely increased left ventricular wall thickness, diastolic dysfunction, and normal systolic function, compatible with infiltrative miocardiopathy.
The patient was treated with prednisone and melphalan for hepatic AL amyloidosis with suspicion of cardiac and renal involvement. After three weeks of treatment, she was admitted for paracentesis because of rapidly developing ascites. She developed a sudden cardiac arrest in the internal medicine ward and died in spite of resuscitations maneuvers. This occurred sixteen weeks after the initial diagnosis. There was no autopsy.
Case 2A 65 year-old man presented a history of upper abdominal discomfort and unquantified weight loss (Table 1). Upper gastrointestinal endoscopy was normal. Symptoms persisted and 3 months later he was admitted to a community hospital with jaundice, dark urine, ascites, hepatomegaly and peripheral edema. Abdominal ultrasound showed an enlarged liver and moderate ascites. He had no history of alcohol or drug abuse. Laboratory tests showed:
- •
ALP 234 U/L (30-100 U/L).
- •
AST 167 U/L (9-25 U/L).
- •
ALT 56 U/L (7-30 U/L).
- •
GGT 203 U/L (4-50 U/L).
- •
Total bilirubin 20.7 mg/dL (0-1 mg/dL).
- •
Albumin 3.5 g/dL (3.5-5 g/dL).
- •
INR 1.7 (0.8-1.2).
- •
Creatinine 1.1 mg/dL (0.5-0.9 mg/dL).
- •
EGFR 71 mL/min/1.73 m2.
- •
Calcium 8.65 mg/dL (8.5-10.5 mg/dL).
- •
Hematocrit 43.3%.
- •
- •
- •
Total urinary protein to creatinine ratio of 2.85.
Serological markers of hepatitis viral infection and autoimmunity were negative. An abdominal CT scan showed hepatomegaly and ascites. The patient developed progressive liver and renal failure (INR 1.9, bilirubin 33 mg/dL, and creatinine 2.3 mg/dL) with grade II hepatic encephalopathy two weeks after the appearance of jaundice. At that moment, the patient was referred to our center with the diagnosis of ALF for an emergency liver transplantation. Upon arrival, the patient deteriorated rapidly, renal failure worsened (creatinine 5.4 mg/dL), he was hypotensive and in shock. The kidneys had a normal size on abdominal ultrasound. Ascitic fluid analysis showed no signs of infection and a total protein concentration 0.7 g/dL and a SAAG of 2.1 g/dL consistent with PH-related ascites.
During the following 36 h, the patient required vasoactive drugs and mechanical ventilation. He presented malignant arrhythmias and finally died in spite of all measures taken in the intensive care unit. A post-mortem liver sample was obtained. Histopathological examination of the biopsy specimen showed amorphous material stained by Congo red. Immunohistiquimical analysis was positive for k light chains compatible with an AL amyloidosis of the liver. During the few hours he stayed in the ICU there was no time for a more complete study, so a plasma cell study was not performed. No autopsy was done.
Case 3A 64 year old man with a history of gout, obesity, hypercholesterolemia and hypertension was admitted with malaise, anorexia, upper abdominal pain and loss of 30 kg in six months (Table 1). Laboratory serum and urine test showed:
- •
ALP 452 U/L.
- •
AST 51 U/L.
- •
ALT 53 U/L.
- •
GGT 341 U/L.
- •
Total bilirubin 1.05 mg/dL.
- •
Prothrombin activity 67%.
- •
Total serum protein 5.6 g/dL.
- •
Serum albumin 3.7 g/dL.
- •
Hematocrit 39%.
- •
- •
- •
Creatinine 1.87 mg/dL (0.5-0.9 mg/dL).
- •
Estimated glomerular filtration rate of 39 mL/min/1.73 m2.
- •
Calcium 9.5 mg/dL.
- •
Urinary protein 180 mg/dL.
- •
Lipase 357 U/L.
Magnetic resonance cholangiography was normal, except for hepatomegaly. Endoscopic ultrasound showed mild amount of sludge in the gallbladder and no stones in the biliary tract. A subsequent abdominal CT scan showed diffuse lytic bone lesions suggestive of multiple myeloma. Serum and urine protein electrophoresis were normal. Immunofixation of immunoglobulins on urine showed an abnormal band which corresponded to a monoclonal immunoglobulin κ light chain component. Bone marrow smear showed 8% of plasma cells. Bone biopsy showed amorphous material that was positive for amyloid with the Congo red stain, and immunorreactivity to light chains (κ and λ). A subcutaneous fat biopsy was performed in search of systemic amyloidosis, with negative results.
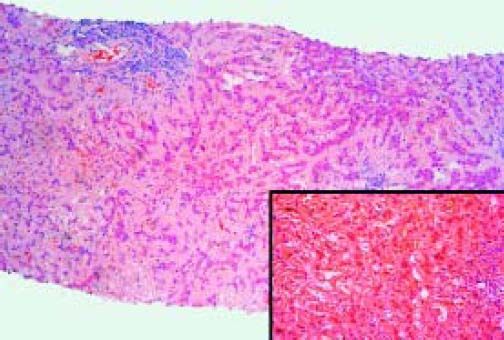
The patient developed fever and back pain, so he was readmitted. The diagnosis was a perivertebral abscess secondary to a methicillin-sensitive Stafilococcus aureus. He underwent surgery for drainage and received antibiotics for several weeks. Liver tests worsened with the following values: ALP 608 U/L, GGT 1027 U/L, total bilirrubin 1.98 mg/dL, AST 122 U/L, ALT 194 U/L and prothrombin activity 89%. He was then evaluated in our Hepatology Unit. At physical examination he had ascites, edema, spider nevi in the chest and abdominal wall collaterals. Markers of viral and autoimmune hepatitis were negative. Diagnostic paracentesis was compatible with PH ascites (total protein concentration 0.8 g/dL, SAAG of 2.4 g/dL). Ascites responded well to diuretic treatment and a liver biopsy was performed (Figure 1). The biopsy showed diffuse deposition by eosinophilic material that was positive for Congo red stain. The material was strongly positive for kappa light chain immunoglobulin, consistent with hepatic amyloidosis type AL. A bone marrow biopsy showed 20% of plasma cells.
. Inset: Congo red staining of amyloid. The material was strongly positive for kappa light chain immunoglobulin (not shown).")
Case 3. Section of the liver shows atrophic liver cell plates and narrow sinusoids by the diffuse deposition of a pale eosinophilic hyaline material in the space of Disse. Lymphocytic infiltration in two portal tracts (Hematoxylin-eosin 80x). Inset: Congo red staining of amyloid. The material was strongly positive for kappa light chain immunoglobulin (not shown).
The patient was evaluated for primary AL amyloidosis therapy, in the context of multiple myeloma. He received prednisone and melphalan. After eight months of therapy, he presented a ventricular arrhythmia that responded to treatment in the emergency room. Echocardiogram showed a modest increase in left ventricular wall thickness, compatible with infiltrative cardiomyopathy, with normal systolic function. The patient was then offered placement of an implantable cardioverter-desfibrillator but he refused this treatment. His renal function worsened in the following months and he needed to start peritoneal dialysis. He died four months after starting dialysis at home. No autopsy was done.
DiscussionAL amyloidosis is one of the most common forms of amyloidosis, and it is thought to be the cause of 1 out of 2,000 deaths in developed nations.2 Its prognosis is related to the involved organs. For instance, cardiac amyloid deposition (as likely happened in patients 1 and 3) and the appearance of congestive cardiac failure confer a median survival of 4-6 months.2
Diagnostic criteria for AL amyloidosis have been developed by the International Myeloma Working Group and require the presence of four elements: an amyloid-related systemic syndrome (e.g. renal, liver, heart, gastrointestinal tract or peripheral nerve involvement); positive amyloid staining by Congo red in any tissue (e.g. fat aspirate, bone marrow or organ biopsy) or the presence of amyloid fibrils on electron microscopy; evidence that the amyloid is light chain related established by direct examination of the amyloid (ideally using mass spectrometry-based proteomic analysis since immunohistochemistry results to type amyloid may be unreliable); and evidence of a monoclonal plasma cell proliferative disorder (e.g. presence of a serum or urine M protein, abnormal serum free light chain ratio, or clonal plasma cells in the bone marrow).8–9
In the three patients reported here, diagnosis of AL amyloidosis was performed in different grounds. Case 1 had cardiac, hepatic and renal syndrome, positive amyloid stain in the liver, evidence that the amyloid was light chain-related in the analysis of the liver biopsy, and evidence of a plasma cell proliferative disorder in the bone marrow (15% plasma cells on bone marrow smear). Case 2 presented progressive liver and renal failure and likely, cardiac involvement. Concerning the study of amyloid, we only have the biopsy criteria in this case since there was no time to do a proper serum free light chain analysis. Case 3 had hepatic, renal and cardiac syndromes, positive stain for amyloid in the liver and bone biopsy, evidence that the amyloid was light chain-related (in both bone and liver biopsy), and plasma cells in the bone marrow (8% on bone marrow smear and 20% in bone marrow biopsy) with an immunofixation on urine positive for monoclonal k light chain band.
Although hepatic biopsy was very useful in our cases, diagnosis of AL amyloidosis with systemic involvement can be made with a subcutaneous fat biopsy or a rectal biopsy in the majority of cases. As a general rule, the less invasive approach must be chosen to obtain a tissue sample in order to minimize the associated risks of during diagnostic work-up. In our cases, diagnosis of hepatic amyloidosis was not suspected prior to histological assessment in cases 1 and 2; in case 3, the diagnosis was suspected and the liver biopsy was done with no complications.
With regard to the hepatic manifestations of our cases, gastrointestinal involvement is common in AL amyloidosis, but severe consequences, such as those described here, are infrequent (Table 2).10 In fact, while liver involvement occurs in around 70% of patients with AL amyloidosis, symptoms derived from this involvement are uncommon.11 Park, et al. published the largest cohort of patients with hepatic AL amyloidosis. In this series, the most common symptoms were weight loss (72%), fatigue (60%) and abdominal pain (53%). Other less common symptoms were edema (26%), anorexia (26%), early satiety (19%), nausea (15%) and dysgeusia (10%). The most common physical finding was hepatomegaly, which was found in 81% of the patients, followed by ascites (42%) and, more infrequently, purpura (15%), splenomegaly (10%) and spider angiomata.11 Finally, the most frequently abnormal blood test was an increased ALP level (86%). In fact, 61% of patients with AL amyloidosis and hepatic involvement had values of 500 U/L or more.
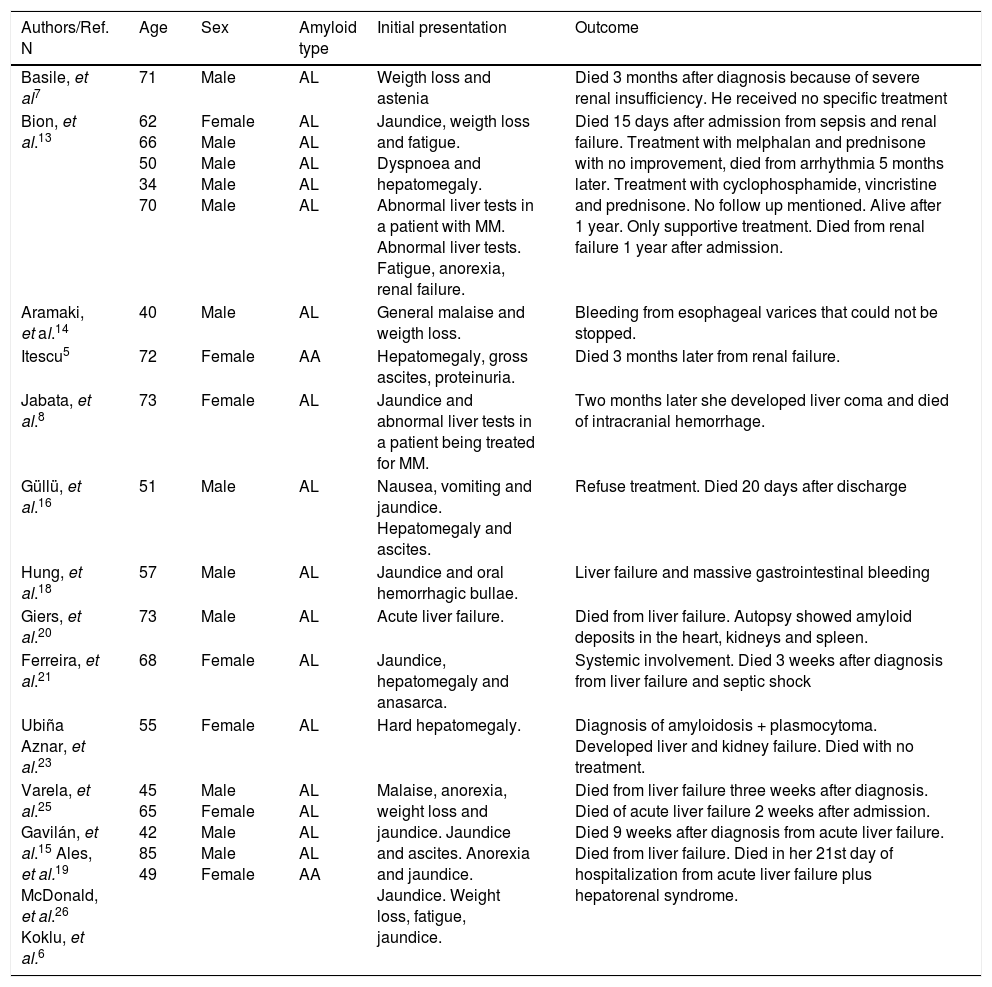
Pertinent literature, (e.g. similar cases of hepatic amyloidosis with portal hypertension or acute liver failure).
| Authors/Ref. N | Age | Sex | Amyloid type | Initial presentation | Outcome |
|---|---|---|---|---|---|
| Basile, et al7 | 71 | Male | AL | Weigth loss and astenia | Died 3 months after diagnosis because of severe renal insufficiency. He received no specific treatment |
| Bion, et al.13 | 62 66 50 34 70 | Female Male Male Male Male | AL AL AL AL AL | Jaundice, weigth loss and fatigue. Dyspnoea and hepatomegaly. Abnormal liver tests in a patient with MM. Abnormal liver tests. Fatigue, anorexia, renal failure. | Died 15 days after admission from sepsis and renal failure. Treatment with melphalan and prednisone with no improvement, died from arrhythmia 5 months later. Treatment with cyclophosphamide, vincristine and prednisone. No follow up mentioned. Alive after 1 year. Only supportive treatment. Died from renal failure 1 year after admission. |
| Aramaki, et al.14 | 40 | Male | AL | General malaise and weigth loss. | Bleeding from esophageal varices that could not be stopped. |
| Itescu5 | 72 | Female | AA | Hepatomegaly, gross ascites, proteinuria. | Died 3 months later from renal failure. |
| Jabata, et al.8 | 73 | Female | AL | Jaundice and abnormal liver tests in a patient being treated for MM. | Two months later she developed liver coma and died of intracranial hemorrhage. |
| Güllü, et al.16 | 51 | Male | AL | Nausea, vomiting and jaundice. Hepatomegaly and ascites. | Refuse treatment. Died 20 days after discharge |
| Hung, et al.18 | 57 | Male | AL | Jaundice and oral hemorrhagic bullae. | Liver failure and massive gastrointestinal bleeding |
| Giers, et al.20 | 73 | Male | AL | Acute liver failure. | Died from liver failure. Autopsy showed amyloid deposits in the heart, kidneys and spleen. |
| Ferreira, et al.21 | 68 | Female | AL | Jaundice, hepatomegaly and anasarca. | Systemic involvement. Died 3 weeks after diagnosis from liver failure and septic shock |
| Ubiña Aznar, et al.23 | 55 | Female | AL | Hard hepatomegaly. | Diagnosis of amyloidosis + plasmocytoma. Developed liver and kidney failure. Died with no treatment. |
| Varela, et al.25 Gavilán, et al.15 Ales, et al.19 McDonald, et al.26 Koklu, et al.6 | 45 65 42 85 49 | Male Female Male Male Female | AL AL AL AL AA | Malaise, anorexia, weight loss and jaundice. Jaundice and ascites. Anorexia and jaundice. Jaundice. Weight loss, fatigue, jaundice. | Died from liver failure three weeks after diagnosis. Died of acute liver failure 2 weeks after admission. Died 9 weeks after diagnosis from acute liver failure. Died from liver failure. Died in her 21st day of hospitalization from acute liver failure plus hepatorenal syndrome. |
PH is an infrequent consequence of hepatic amyloidosis. Ascites is a common feature and is usually associated to a high SAAG.10 In our three cases, ascites was present with a SAAG compatible with PH ascites. Though it is very possible that they all had cardiac amyloidosis, the ascites in all three cases had low protein level, which is not characteristic of cardiac ascites.10 The magnitude of PH seems to be related to the degree of hepatic infiltration. In AL amyloidosis, amyloid is predominantly deposited in Disse’s space, thus reducing the sinusoidal lumen and increasing its resistance to blood flow.11,13 In secondary amyloidosis, the abnormal protein is deposited mainly on the walls of blood vessels.13 PH has important implications in patients with AL as has the potential of developing severe complications, as in the case of cirrhotic patients.10,13 The diagnosis of PH is useful for recognizing and treating its complications, especially gastro-esophageal variceal bleeding.14 Even though the presence of ascites is an important clue, it is noticeable that in one of our patients, it was accompanied by the presence of collateral circulation (visible at physical examination), which made PH easier to suspect.
Liver failure is another uncommon manifestation of AL amyloidosis and usually presents itself as a severe and progressive intrahepatic cholestasis.15,16,26 However, sometimes the disease may be associated to a rapid development of liver failure fulfilling the criteria of ALF (defined by coagulation abnormality plus encephalopathy), as we described in case 2.17–20,22,23–25,27 ALF is a very rare and severe manifestation of AL amyloidosis and emergency liver transplantation has been described as a life-saving procedure in this setting.22,28 It is unknown why some cases of AL hepatic amyloidosis manifest as ALF and not as a slowly progressive disease. In some cases a triggering event can be identified (infection or spontaneous spleen rupture), however this is not always the case. Although our Case 2 presented as ALF, the patient’s symptoms started several months before presentation mainly with weight loss and malaise, thus, ALF seems to be the final stage of a longer disease. Noteworthy, the 2011 American Association for the Study of the Liver guidelines state that ALF may occur in a patient with an already established liver disease if the diagnosis has less than 26 weeks, as it can occur in Wilson’s disease, autoimmune hepatitis and vertically-acquired hepatitis B virus infection.29 We think that these criteria can be applied in case 2. It is also worth noting that this case presented PH (ascites with a high SAAG) as a concomitant manifestation of hepatic amyloidosis and that PH and concomitant ALF are very uncommon manifestations of amyloidosis.6,15
Liver involvement in AL amyloidosis determines a poor prognosis30 with a median survival of 8.5 months; predictors associated with survival are the presence of congestive heart failure at the moment of the diagnosis of liver amyloidosis, a platelet count over 500 x 109/L, and a serum total bilirubin over 20 mg/dL.
Therapy of primary amyloidosis is still associated to a relatively poor success rates although the therapeutic arsenal for the management has been considerably enriched in the last decade. For most experts, the mainstay of the treatment of primary amyloidosis is stem cell transplantation (SCT). However, only a minority of the patients is eligible, and this procedure is not free of morbidity and mortality. For those who are not proper candidates for SCT, melphalan and steroid-based therapy is the option for virtually all patients but the response rate to this regimen is unsatisfactory and the impact on survival discrete.31 Although SCT has not been demonstrated to have a survival advantage compared to chemotherapy, recent evidence suggests that a strict selection of patients allows reducing SCT related morbidity and mortality.32 Lenalidomide, pomalidomide, bortezomib and the combination of thalidomide with agents such as melphalan, steroids and cyclophosphamide are being studied as alternative regimes mostly for those who are ineligible or fail to improve after SCT.31
ConclusionsHepatic involvement of AL amyloidosis is not an uncommon condition, it is associated with reduced survival but rarely has clinical manifestations. PH is an infrequent finding in AL amyloidosis and can be clinically manifested by the presence of gastroesophageal varices (and eventually variceal bleeding) and ascites. Liver failure can be progressive and, in some very unusual cases, it may present as ALF. A high clinical suspicion is important to establish a correct diagnosis.
Abbreviations- •
ALF: acute liver failure.
- •
ALP: alkaline phophatase.
- •
ALT: alanine aminotransferase.
- •
AMA: antimitochondrial antibody.
- •
ANA: antinuclear antibody.
- •
AST: aspartate aminotransferase.
- •
EGFR: estimated glomerular filtration rate.
- •
ESR: erythrocyte sedimentation rate.
- •
GGT: gammaglutamyltransferase.
- •
Ig: immunoglobulin.
- •
INR: international normalized ratio.
- •
PH: portal hypertension.
- •
SAAG: serum to ascites albumin gradient.
- •
SMA: smooth muscle antibody.
This paper was partially supported by grants from the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) #11100113 (to CB) and • 110455 (to MA).



